Chapter 5 Infectious diseases
5.1 Viral, mycoplasmal and rickettsial infections
Viral infections
Many viruses infect the lower respiratory tract. They include the orthomyxoviruses (influenza virus), paramyxoviruses (parainfluenza viruses, measles virus and respiratory syncytial virus), adenoviruses, herpesviruses (cytomegalovirus, varicella-zoster virus and herpes simplex virus) and formerly variola virus (smallpox). Many of these viruses are of course also responsible for non-respiratory disease. The role of papillomavirus in neoplasia of the respiratory tract is discussed on page 535 and parvovirus is mentioned as a cause of hydrops fetalis on page 43. The role of herpes-like viruses in Kaposi’s sarcoma and body cavity-based lymphomas is discussed on pages 635 and 734 respectively.
Viral infection of the lower respiratory tract occurs in three general situations: infections confined to the respiratory tract, systemic infections that involve the lung and opportunistic infection of the lungs in the immunocompromised (Box 5.1.1).
Respiratory viruses may strike any part or all the respiratory tract but on the whole each tends to affect particular parts and thereby elicit characteristic clinical effects (Fig. 5.1.1). This pattern varies however if there is some predisposing cause. Thus, the rhinoviruses, which usually cause nothing more serious than a cold, are the commonest viral trigger of acute exacerbations of chronic bronchitis, while in the immunodeficient herpes simplex virus and cytomegalovirus are serious pathogens in the lower respiratory tract.
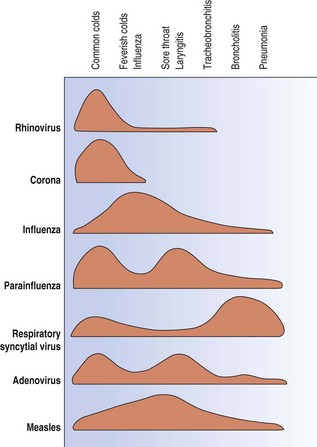
The frequency of these various infections also differs in children and adults. In children, respiratory syncytial virus is the most important viral cause of lower respiratory tract disease, typically causing an obstructive bronchiolitis. Parainfluenza viruses are the most frequent cause of viral pneumonia in children, and the influenza virus in adults. These are followed by the measles and adenoviruses in children and varicella virus in adults, whilst the immunocompromised are also susceptible to cytomegalovirus and herpes simplex virus. A viral aetiology is responsible in 7–15% of adults admitted to hospital with community-acquired pneumonia (see Table 5.2.2, p. 178).
The virus responsible can be cultured from sputum or nasopharyngeal washings and evidence of infection by particular viruses may be provided by the demonstration of a rising titre of specific antibodies in the patient’s serum. The advent of monoclonal antibodies has provided specific sensitive and reproducible probes that can be directly conjugated to a fluorescent tag so that the examination of exfoliated cells for viral antigen by immunofluorescent techniques is now the method of choice for the rapid identification of most respiratory viruses. In tissue, the particular virus may be identified by immunocytochemistry, electron microscopy or gene probes.1–7
The pathology is modified to some extent by the type of virus responsible but infections caused by different viruses have many features in common.8 In the lungs, as in other organs, some viruses have a cytopathic effect and kill the infected host cells, whilst others stimulate proliferative activity. Thus, influenza, adenovirus, varicella and herpes simplex pneumonia are all characterised by epithelial cell necrosis (Figs 5.1.2 and 5.1.3), whilst respiratory syncytial virus and measles virus stimulate mitotic division and cause characteristic proliferative changes in the bronchioles and alveoli respectively. The distribution of the changes is often characteristic of a particular virus, but not specific. Thus, within the alveoli, influenza tends to affect the epithelium diffusely. The effects of adenovirus, on the other hand, are generally maximal in the region of the terminal bronchioles, whilst varicella pneumonia is also focal but lacks any particular relationship to the acinar architecture. Viral inclusions are evident in certain viral pneumonias, notably measles, adenovirus, cytomegalovirus, varicella and herpes simplex pneumonia, but not in others.
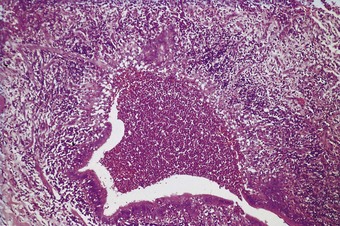
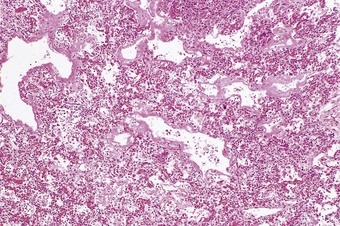
Figure 5.1.3 Viral pneumonia causing necrosis of the alveolar epithelium with the formation of hyaline membranes. The virus responsible in this patient was that of measles but many respiratory viruses have a similar effect, as do several other cytotoxic factors: see diffuse alveolar damage, in Chapter 4.
(Courtesy of Dr V Chrystal, Durban, South Africa.)
Alveolar epithelial necrosis is a feature of severe viral pneumonia. It causes the formation of hyaline membranes (Fig. 5.1.3) and the pathology of such viral pneumonia is essentially that of diffuse alveolar damage (see Chapter 4). The regenerative changes seen in diffuse alveolar damage may be evident, possibly involving epithelial metaplasia.
Influenza
Microbiology and epidemiology
Influenza is epidemic almost annually in the winter months in many parts of the world and at long intervals the disease occurs in pandemic form, notably in 1889–92, 1918–19, 1957–58, 1968 and 2009. It is estimated that in the pandemic that followed the world war of 1914–18, some 20–30 million people died from the disease in little more than a year, more than were killed in the war itself. Until 1933 it was widely believed that the disease was caused by the bacterium Haemophilus influenzae. The discovery in 1933 that the disease could be transmitted to ferrets by intranasal inoculation of filtered washings from the noses of patients established its viral nature.9
There are several types of influenza virus. All are originally bird viruses, some crossing from birds to species such as humans, pigs, horses and seals. Once established in the new species they undergo constant changes in their antigenic makeup, a feature that necessitates the constant manufacture of new vaccines. The strains involved in most serious human infections belong to types A and B, with the former much the more virulent. Outbreaks of infection with influenza A occur most years, with epidemics every 5–15 years. Influenza B also causes epidemics, but less frequently. Influenza C does not appear to cause epidemics. Successive epidemics appear to be decreasing in severity, possibily due to natural selection involving evolutionary changes that favour transmissibility over pathogenicity.10
The haemagglutinin molecule enables the virus to attach to host cells prior to infecting them and neuraminidase permits new virus particles to bud from the cell membrane. Although there are considerable differences between the influenza viruses that infect different species, the haemagglutinin molecule’s lability occasionally permits its virus to switch from one host species to another, in which the disease is likely to spread in epidemic form. It appears that all the devastating influenza pandemics of the twentieth century, such as Spanish flu in 1918, Asian flu in 1957 and Hong Kong flu in 1968, were caused by viruses that made such a switch from birds to humans.11
Recent genomic studies of the H1N1 virus responsible for the 1918 pandemic suggest that it was an avian virus that adapted to humans, rather than developing by a reassortment of avian and human viruses, as in the 1957 and 1968 pandemics.12
The switch from animals to humans in the ‘wet markets’ of the Far East is also relevant to the severe acute respiratory syndrome (SARS), described on page 163.
In 2004, the H5N1 strain of the influenza A virus devastated flocks of poultry in Asia and was responsible for the deaths of some humans from what was termed ‘Asian bird flu’.13,14 This strain was not very infective to humans, possibly because the temperature of the human nose (32°C) is too cold for a virus whose natural host is the avian intestine (temperature 40°C).15,16 However, when the virus succeeded in infecting humans it was particularly virulent for two reasons. First, it was resistant to the antiviral effects of human interferons and tumour necrosis factor-α, a property acquired by a simple mutation in the non-structural gene resulting in a change from aspartate to glutamate at position 92.17 Second, it preferentially targeted the alveolar rather than the tracheobronchial epithelium.18
The year 2009 saw swine influenza A/H1N1 virus switch to humans and exhibit the ability to spread from case to case, rapidly assuming pandemic proportions. Pigs are unusual in that their respiratory epithelium has receptors for both avian and human influenza viruses so that they can be infected with these viruses simultaneously, providing the ideal environment for genetic reassortment. This appears to have taken place as the 2009 swine influenza virus shows a novel reassortment of genes derived from swine, avian and human influenza viruses, a feature that is probably responsible for this virus being able to infect both pigs and humans. Unlike seasonal influenza, the disease made its first appearance in the northern hemisphere, apparently originating in Mexico, and its first impact was during the summer months. There was much mild disease but again, in contrast to seasonal influenza, severe respiratory failure was seen in young, previously healthy persons, as is often the case in influenza pandemics. This feature is not well explained but it is possible that the healthy mount a more vigorous immune response that is in some way detrimental to the host. Older people were less susceptible but more likely to die when infected.19 However, one year later it was apparent that the pandemic had not proved as severe as first thought.
Clinical features
Primary influenzal pneumonia is generally rare but figured prominently in the 1918–19 pandemic. It carries a high case fatality rate and in the 1918–19 pandemic it was notable that mortality was highest in adults aged 25–34, possibly because older people had been exposed earlier to a similar strain of the virus.20,21 Primary influenzal pneumonia may be fulminant, leading to the death of a previously healthy person within a few hours of the onset of symptoms.22
Pathology of uncomplicated influenza
The cytopathic effect of influenza virus is seen microscopically in characteristic degenerative changes in the epithelial cells of the bronchial and bronchiolar mucosa. These changes involve all cells of the surface epithelium and often the cells lining the bronchial glands: swelling of the cells, vacuolation of their cytoplasm and degeneration of the nucleus proceed to cell loss and frank necrosis (Fig. 5.1.4). Viral inclusions are not evident but the virus can be identified in tissue sections by immunocytochemistry and in situ hybridisation.23,24 The deeper tissues show oedema, hyperaemia and a moderate to marked accumulation of lymphocytes; neutrophils are present but account for only a small proportion of the cellular infiltrate.
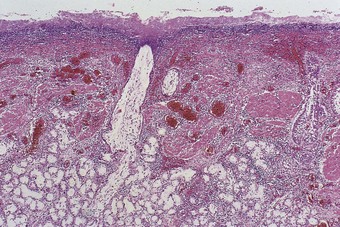
Alveolar involvement is unusual but cases of fulminating influenzal viral pneumonia are occasionally encountered, particularly with the 1918 and more recent H5N1 and H1N1 strains.14,24,25 Autopsy in such cases shows that the lungs are bulky and deeply congested.14,22,24a Blood-stained, frothy fluid oozes freely from the cut surface. Areas of haemorrhage are present and may be extensive. The mucosa of the bronchial tree is very hyperaemic. Microscopically, the alveoli contain a fibrin-rich oedema fluid, which is often frankly haemorrhagic. Macrophages may be numerous in the exudate and hyaline membranes are often found lining the alveoli. T lymphocytes are often prominent in the alveolar interstitium while focal necrosis of alveolar walls and thrombosis of capillaries are conspicuous features in the parts most severely affected.26
Postmortem examination of two victims of the H5N1 virus showed similar evidence of diffuse alveolar damage but it was also found that viral replication in the respiratory tract had resulted in particularly high levels of cytokines such as interferons and tumour necrosis factor-α, which, with the resultant haemophagocytic syndrome, was considered to be the chief cause of death.27 Generally, however, H5N1 viral pneumonia shows no special features and it may be difficult to distinguish the changes brought about by the H5N1 virus from those caused by viruses such as that responsible for SARS or by factors such as acid aspiration or oxygen toxicity. More specific tests such as viral isolation, in situ hybridisation and reverse transcription-polymerase chain reaction (RT-PCR) are required to confirm H5N1 infection.
Fulminant uncomplicated influenzal pneumonia is often associated with changes in other parts of the body that indicate the occurrence of influenzal viraemia14: among these are haemorrhagic encephalomyelitis, which is an acute infective condition distinct from postinfluenzal encephalomyelopathy, rhabdomyolysis and placentitis.28,29
Bacterial superinfection in influenza
Although pneumonia is the usual cause of death in epidemics of influenza, it is often a secondary bacterial pneumonia that is responsible. Neutrophilic exudates, organising pneumonia and bronchiolitis obliterans are then added to or replace the changes seen in uncomplicated cases.22,24,30–33 The impact an influenza epidemic has on respiratory death rates in general is shown in Figure 5.1.5.
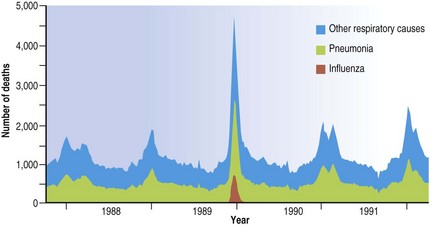
Before the discovery of the influenza virus in 1933, the changes that are now known to be due to the viral infection were often confused with those of complicating infections, mainly caused by bacteria. Haemophilus influenzae was first isolated in the 1889–90 pandemic and so named because its discoverer, Pfeiffer, recovered it from a large proportion of cases and mistook it for the cause of influenza. In the 1918–19 pandemic H. influenzae was again found, along with Streptococcus pneumoniae and Staphylococcus aureus.32 With the advent of antibiotics, resistant strains of Staphylococcus aureus emerged and in the 1957–58 pandemic staphylococcal superinfection of the lungs was the major fatal complication of influenza. In the 1968 epidemic Streptococcus pneumoniae was the principal bacterial pathogen in the elderly and Staphylococcus aureus in the young.30
The relationship of the staphylococcus and the influenza virus has been much studied and there is evidence that each promotes the growth of the other.34 Thus, certain staphylococci have a protein in their cell wall that binds to the Fc region of immunoglobulin G. In the presence of anti-influenzal serum this protein enhances staphylococcal binding to cells infected by the influenza virus35 and in this way the staphylococcus takes advantage of the host’s immune reaction to the influenza virus. In turn, the staphylococcus aids entry of the virus into the host’s cell. It does this by secreting a protease that activates a viral surface protein necessary for penetration of the host’s cells36: normally such proteases are in short supply, so limiting the rate at which influenza virus can infect cells and reproduce. The relationship of influenza and Streptococcus pneumoniae infection has been studied in less detail but there is evidence of similar enhancement of bacterial adherence to tracheal epithelium following influenza infection.37
Parainfluenza
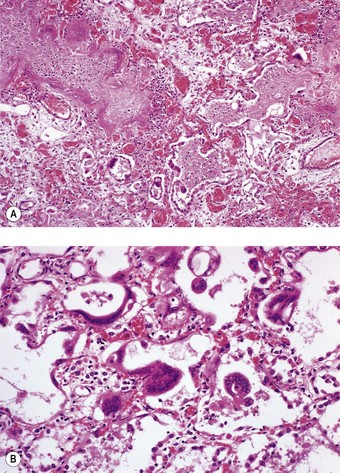
Parainfluenza is caused by the parainfluenza viruses, not by Haemophilus parainfluenzae. It is commoner in children and particularly affects the larynx, causing croup. There may be necrosis of the mucosa as in influenza (Fig. 5.1.6A), and quite frequently small polypoid growths of the bronchial and bronchiolar epithelium develop, similar to those associated with infection by respiratory syncytial virus (see below). In the lung there is hyperplasia of the alveolar epithelium and a serous exudate containing increased numbers of macrophages is seen. In immunosuppressed individuals, parainfluenza type III virus may result in a giant cell pneumonia that is indistinguishable from that of measles except that the inclusion bodies typical of measles pneumonia are not a feature (Fig. 5.1.6B).38–41
Respiratory syncytial virus
Epidemiology
Respiratory syncytial virus was first isolated in 1956 from an outbreak of coryza in a colony of chimpanzees and its infectivity for Man was shown when one of the investigators of this epizootic illness contracted the disease. It has since been shown that the virus frequently infects the lower respiratory passages of Man, particularly the young.42 Specific neutralising antibodies indicative of an earlier infection are found in the serum of almost all children over the age of 5 years in Britain. Most children merely develop a cold but a few suffer from severe bronchiolitis, which in the developing world is an important cause of death in the very young.42a,b Those that recover are prone to develop recurrent wheeze later in childhood.42c The immunity infection conveys is incomplete and reinfections may occur throughout life.
Respiratory syncytial virus infection shows a marked seasonal pattern, producing annual epidemics each winter in temperate climates and in the hot rainy season in tropical countries. During an incubation period of 3–6 days the virus replicates in the upper respiratory tract, causing fever, cough and coryza. Spread from the upper to the lower respiratory tract may occur, with consequent bronchiolitis and pneumonia.
Particularly important is the bronchiolitis that respiratory syncytial virus is prone to cause in infants. The conductive airways of small infants are quite narrow and easily blocked by relatively small amounts of inflammatory exudate: because of this, fatal asphyxia is liable to follow respiratory syncytial virus infection. This is particularly the case in those who have airflow obstruction as a consequence of prematurity and its treatment. Several epidemics of acute bronchiolitis due to this virus have been described among infants. These outbreaks are often remarkably focal in distribution, affecting only a comparatively small area or community. Infants with bronchopulmonary dysplasia and those who have congenital heart disease or are immunocompromised are particularly at risk and units specialising in these underlying conditions have to guard against nosocomial spread of infection.43–45 However, most children with respiratory syncytial virus infection have no predisposing factors and even previously healthy children may suffer fatal infection.42,46
Pathogenesis
It is notable that infants under six months of age are particularly prone to respiratory syncytial virus infection. Although this is a period when the infant benefits from the presence of maternal antibodies, placental antibody transmission is selective, being better for immunoglobulin G than A. Breast-feeding protects against respiratory syncytial virus infection,47 presumably by virtue of breast milk being rich in immunoglobulin A. It has been noted that infants immunized against the virus and subsequently infected naturally, suffer a more severe illness than those not so immunised,48 suggesting that the damage is mediated immunologically.49,50 This is supported by the finding that the typical bronchiolitis is characterised by scanty virus whereas in the rarer pneumonic form of the disease the virus is abundant. This is compatible with the bronchiolitis representing an adverse immune reaction and the pneumonia being the result of direct viral damage to the lungs.51 The cytokine profile suggests that the reaction involved in the bronchiolitis involves a predominantly type 2 response characterised by high interleukin-10/interleukin-12 and interleukin-4/γ-interferon ratios.52 Suspicion has fallen upon the formaldehyde inactivation of the vaccine creating reactive carbonyl groups on the antigen.52a Passive immunisation, conferred by monthly injection, is free of the hazards induced by active immunisation and is recommended for high-risk babies.53
Histopathology
The virus infects the bronchiolar epithelium and usually leads to its destruction.54,55 Occasionally cytoplasmic inclusion bodies may be seen in degenerating bronchiolar epithelial cells or the virus may be demonstrated by immunocytochemistry.1,55 Regeneration involves the proliferation of poorly differentiated cells which form a stratified non-ciliated epithelium. Occasionally micropolypoid epithelial protrusions are evident (Fig. 5.1.7).8 The bronchioles are occluded by plugs of mucus, fibrin and epithelial cell debris, and cuffed by an infiltrate of lymphocytes, plasma cells and histiocytes. Except in the immediate vicinity of the bronchioles, alveoli are generally not involved in the inflammatory process. If, however, infection is on a major scale there may be pneumonia with the general features of a viral pneumonia, as described above.46 In severe immunodeficiency, respiratory syncytial virus may cause giant cell pneumonia,56 a condition that is more often caused by measles virus.
Metapneumovirus
Metapneumovirus was only discovered in 200157 but is now recognised to be ubiquitous; serological evidence of past infection is universal by the age of 5 years. Much of this goes unrecognised but metapneumovirus infection is nevertheless a leading cause of lower respiratory tract illness in young children. In one investigation the virus or its DNA was found in 20% of nasal-wash specimens previously declared virus-negative that had been collected from otherwise healthy infants and children suffering from a lower respiratory tract illness: the infection was associated with bronchiolitis in 59% of cases, pneumonia in 8%, croup in 18% and exacerbation of asthma in 14%, a spectrum of disease similar to that found with respiratory syncytial virus.58 The pathological changes are not well described.
Measles
Clinical features and epidemiology
Measles has been regarded as one of the inevitable infections of childhood but with an effective safe vaccine now available this is no longer necessary (Fig. 5.1.8).59 In the developed countries first and more recently in the developing world, immunisation against measles has been promoted vigorously, with spectacular success. Between 2000 and 2007 mortality from measles fell by 74% worldwide and by 89% in Africa, where it was formerly a major cause of death in childhood. The Americas were declared free of endemic measles transmission in 2002 and cases there now occur only as a result of its importation from other countries.60,61 The disease has not yet been eradicated in the UK, partly because unfounded claims that the vaccine is responsible for autism led to declining vaccine uptake with consequent focal outbreaks of measles.62 Other European countries still experiencing large outbreaks include the Ukraine, Switzerland and Austria.
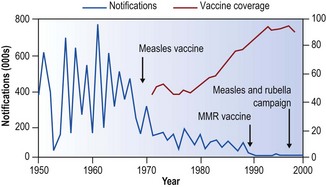
Figure 5.1.8 Annual measles notifications and vaccine coverage in England and Wales 1950–1999.
(Adapted by permission from BMJ Publishing Group Limited.56)
Where measles is prevalent, the incidence is usually highest in the early spring, when droplet infections are particularly rife. The epidemics have a remarkably consistent biennial character (see Fig. 5.1.8) and the explanation of this has been the subject of several interesting hypotheses. The one most favoured envisages waning immunity over the succeeding 2 years in those children who had only a subclinical illness in the last epidemic. With the influx of two further entries into infant schools, a new population of susceptible children is formed that is liable to contract overt disease when the seasonal conditions are again favourable for the spread of the virus. In this way a fresh epidemic develops. Overt clinical disease, on the other hand, confers lifelong immunity.
Pneumonia is a rare complication of measles in the western world but is common in malnourished African children, in whom it frequently proves fatal.63 That pneumonic foci may develop in prosperous countries in the course of severe but non-fatal attacks of measles is shown by the demonstration in some such cases of patchy opacities on chest radiography; in almost all these cases the condition resolves rapidly. The cause is usually one of the common bacterial pathogens but it can be another virus taking advantage of the patient’s debility and impaired cellular immunity. Measles virus infection is characterised by both the development of a strong antiviral immune response and abnormalities of immune regulation: there is often a poor skin response to common antigens and helper/suppressor T-cell ratios may be low in both the blood and bronchoalveolar lavage, suggesting that cellular immunity is impaired.64 Thus, measles has predisposed to both adenovirus and herpesvirus pneumonia.65,66 Secondary pulmonary infection is responsible for about half the mortality in measles.67 Other causes of death include measles pneumonia and measles encephalitis.
Measles may also be very severe when it affects immunodeficient patients, whether they are suffering from primary immunological defects, acquired diseases such as leukaemia or conditions which require treatment with cytotoxic or immunosuppressant drugs.68 Such patients may have unpredictable responses to measles virus. They may have a rash but fail to produce antibodies, or they may fail to develop a rash although infected with the virus. Fatal measles pneumonia in a previously healthy adult is very rare.69
Pathology of measles pneumonia70,71
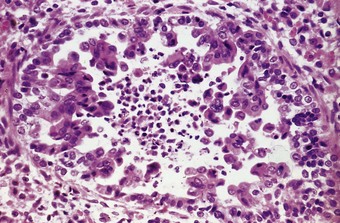
Microscopically, there are degenerative changes in the epithelium of the bronchi and bronchioles, often accompanied by hyperplasia, particularly in the small airways. As in influenzal pneumonia, squamous metaplasia may occur and mitotic figures may be numerous. Measles pneumonia may take the form of diffuse alveolar damage with hyaline membrane formation (see Fig. 5.1.3), or, more characteristically, multinucleate giant cells may line the alveolar ducts and alveoli (Fig. 5.1.9). Electron microscopy shows that the giant cells are formed from type II alveolar epithelial cells.71,72 The giant cells contain prominent cytoplasmic and nuclear viral inclusion bodies that are clearly evident in eosin-stained sections. Being epithelial, the pulmonary giant cells are quite different from the Warthin–Finkeldey giant cells that are found in lymphoid tissue throughout the body in measles, particularly in the immediately pre-exanthematous stage.
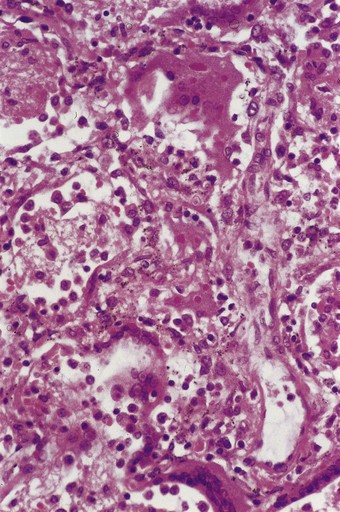
Figure 5.1.9 Measles giant cell pneumonia. There is a syncytial proliferation of type II pneumocytes containing eosinophilic cytoplasmic and nuclear viral inclusions. This response is typical of measles pneumonia but is occasionally encountered with other forms of viral pneumonia (see Fig. 5.1.6).
Differential diagnosis
Measles is the commonest cause of giant cell pneumonia but occasionally other viruses such as parainfluenza, respiratory syncytial and varicella-zoster viruses are responsible, especially in the immunocompromised (see Fig. 5.1.6).38,56,73 The diagnosis is usually evident clinically or is made serologically but immunohistochemistry can be performed on tissue sections. Hard-metal disease is also characterised by the presence of alveolar epithelial polykaryons but these are more focal than those of measles, lack viral inclusion bodies and are not accompanied by such severe interstitial pneumonia (see Fig.7.1.25, p. 354).
Adenovirus
Adenovirus pneumonia occurs sporadically and in epidemics, particularly in children and young adults, and occasionally complicates measles.65,66 Adenovirus pneumonia is usually combined with bronchiolitis and the lesions are most severe at the centres of the acini, being concentrated on the bronchioles. The virus causes necrosis of the bronchioles, many of which are totally destroyed or are recognisable only by their muscle coat: hyaline membranes replace the necrotic epithelium (Fig. 5.1.10A). Surviving epithelial cells show nuclear inclusions of varying staining reaction: some are diffusely basophilic or amphiphilic and fill the entire nucleus apart from a rim of chromatin (Cowdry type B) while others are eosinophilic and surrounded by a clear halo (Cowdry type A). The bronchioles are cuffed by a lymphoid infiltrate and may show proliferative epithelial activity, variously interpreted as being the result of viral stimulation or of regeneration.54,74 The alveolar tissue shows a mononuclear interstitial pneumonia.
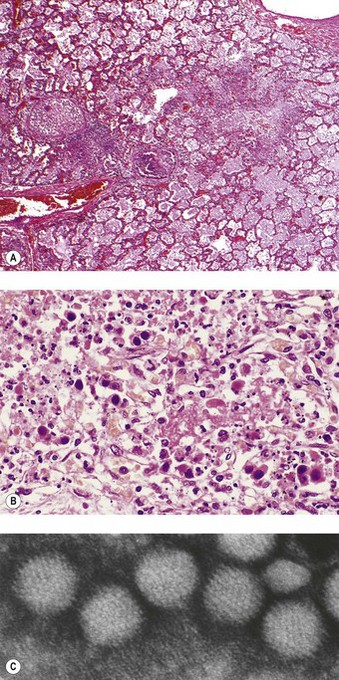
The intranuclear viral inclusions measure up to 5 µm and eventually disrupt the nucleus, leaving so-called smudge cells (Fig. 5.1.10B, C). Healing may be by complete resolution or the pneumonia may be complicated by bronchiolitis obliterans or bronchiectasis.75
Severe acute respiratory syndrome
SARS first appeared in southern China in 2002, from where it quickly traversed the globe, facilitated by air travel and coming to international attention particularly after an outbreak in Hong Kong in 2003.76–81 Cases were subsequently identified in many countries and about 10% of those affected died.82–85 The cause was a previously unknown coronavirus that had switched from civet cats encountered in Asian food markets and adapted to human transmission.86,87 Transmission is air-borne but requires close person-to-person contact. There is no evidence of transmission following casual contact. The virus has subsequently been identified in Chinese horseshoe bats and it is likely that these animals represent the natural reservoir of the virus, with the civets merely acting as carriers.88,89
Clinical features
The incubation period ranges from 1 to 10 days, following which there is a prodromal fever, cough and dyspnoea. Less common symptoms include headache, diarrhoea, dizziness, myalgia, chills, nausea, vomiting and rigor.90 There is no apparent sex predilection and the age distribution is wide. Common laboratory features include lymphopenia involving both CD4 and CD8 lymphocytes, thrombocytopenia, prolonged thromboplastin time, elevated alanine transminase, lactate dehydrogenase and creatinine kinase. Positive viral recovery rates from urine, nasopharyngeal aspirate and stool specimen have been reported to be 42%, 68% and 97% respectively on day 14 of illness, whereas serological confirmation may take 28 days to reach a detection rate above 90%. However, quantitative measurement of blood SARS coronavirus RNA using real-time RT-PCR techniques has a detection rate of 80% as early as day 1 of hospital admission.91
Radiographic abnormalities include focal, multifocal or diffuse opacities. Computed tomography is more sensitive, sometimes showing extensive consolidation in patients with normal chest radiographs. However, the radiological features are not specific and need to be correlated with the clinical and histological findings.92
Pathogenesis
Although pulmonary involvement is the dominant clinical manifestation, extrapulmonary features are common and the virus can often be recovered from faeces and urine, indicating that it is widely distributed in the body. The identification of a specific receptor for the virus is relevant to its tissue distribution. The receptor, a metallopeptidase known as angiotensin-converting enzyme 2, is expressed particularly strongly by pulmonary alveolar and small intestinal epithelia and vascular endothelia.93–95
Pathology
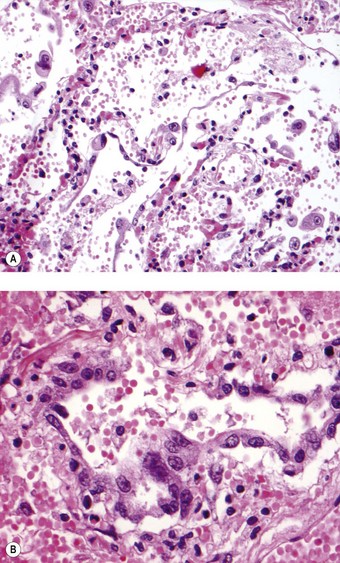
The histology varies according to the duration of illness but the predominant pattern is diffuse alveolar damage.96–100 Cases of less than 10 days’ duration show air space oedema and hyaline membranes whereas those of longer duration exhibit type II pneumocyte hyperplasia, squamous metaplasia, multinucleated giant cells and acute bronchopneumonia succeeded by intra-alveolar organisation.26,96 The alveolar pneumocytes may also show striking cytomegaly with granular amphophilic cytoplasm (Fig. 5.1.11), that sometimes contains eosinophilic inclusions akin to the Mallory bodies of alcoholic hepatitis.96,97 Less common features include haemophagocytosis and thrombosis.97,99 The virus can be identified by RT-PCR in fresh or formalin-fixed, paraffin-embedded lung tissue. Electron microscopy may reveal the viral particles in the cytoplasm of epithelial cells.97–99
Treatment and prognosis
Treatment with corticosteroids, broad-spectrum antibiotics and antiviral agents has been beneficial.98,102 Interferon-α may also have a role.103 However, infection control is as important as pharmacological therapy in this disease.
In the acute phase, SARS is associated with considerable morbidity and mortality, with a global case fatality rate ranging from 7 to 27% (average about 11%). Adverse prognostic factors include advanced age, coexistent disease, high lactose dehydrogenase levels and high initial neutrophil counts. CT data on the extent of the disease are also useful in assessing prognosis.104 Clinical follow-up of patients who recover has demonstrated residual abnormalities of varying degree, including abnormal lung function and patchy fibrosis.105,106 Many survivors also experienced transient pituitary dysfunction.107
Herpes simplex108,109
Virology and predisposing causes
Herpes simplex virus typically causes mucocutaneous vesiculation, serotype 1 (HSV1) generally affecting the oronasal area and serotype 2 (HSV2) the genital mucosae. As with all herpesviruses, infection is lifelong; the virus remains dormant until immunity weakens, which may be triggered by a variety of factors. In these circumstances either serotype may involve the lower respiratory tract. Respiratory infection by HSV1 is more commonly encountered in adults, the infection spreading from the oropharynx, whereas neonatal respiratory infection is usually due to HSV2 as a component of generalised haematogenous disease.110,111 Infection of the lower respiratory tract takes the form of tracheobronchitis or pneumonia. Herpes simplex tracheobronchitis is predisposed to by damage to the respiratory epithelium, especially factors that lead to squamous metaplasia, such as endotracheal intubation and burns.112–116 Risk factors for herpes simplex pneumonia include transplantation,117 cytotoxic chemotherapy and human immunodeficiency virus (HIV) infection118: herpes simplex pneumonia is rare in the immunocompetent.119
Pathology
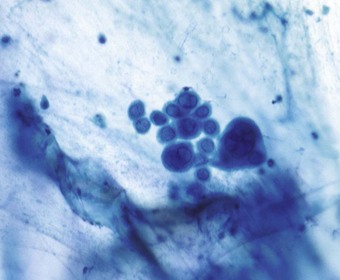
In herpes simplex tracheobronchitis there is extensive mucosal ulceration and pseudomembrane formation. Viral inclusions are most prominent at the periphery of the ulcers and may be identified in exfoliated cells (Fig. 5.1.12). If they are not well developed an immunostain may establish the diagnosis. Long-standing airway infection leads to luminal narrowing and obstructive features. In the lungs the changes are very similar to those of adenovirus pneumonia, including the presence of Cowdry type B ground-glass intranuclear viral inclusions, although these are more eosinophilic than those of adenovirus. Both adenovirus and herpes simplex pneumonia bear a superficial resemblance to bacterial bronchopneumonia but the similarity is in the distribution of the lesions rather than their character: the bronchioles and centriacinar alveoli are mainly affected but the lesions are characterised by necrosis and the accumulation of nuclear debris rather than exudation of neutrophils. Occasionally herpes simplex infection takes the form of a focal necrotising pneumonia more typical of varicella infection (see below). Alternatively there may be arterial involvement in herpes simplex pneumonia with a necrotising vasculitis affecting small and medium-sized pulmonary arteries.120 Sometimes the pneumonia is diffuse: it is suggested that focal disease represents extension of oral mucocutaneous herpesvirus infection down the tracheobronchial tree into the lung whereas diffuse pneumonia is the result of haematogenous spread.108
Cytomegalovirus
Virology and epidemiology
Cytomegalovirus is a serious pathogen in transplantation recipients,121 possibly because the virus replicates best in cells that are activated, as in a transplanted organ. The risk is greatest with bone marrow transplantation, intermediate with heart, lung and liver transplantation and lowest with renal transplantation. However, donor and recipient matching for cytomegalovirus status has reduced the incidence of transmission from the donor. Before the introduction of this policy, fatal cytomegalovirus pneumonia or systemic infection was common. Today reactivation of latent infection is a more common problem but it is important to distinguish the mere presence of viral inclusions from pneumonitis. When a lymphoid infiltrate accompanies the viral inclusions it is also important to distinguish an infective pneumonitis from lung allograft rejection: generally, the infiltrate of cytomegalovirus pneumonia lacks the perivascular lymphocyte distribution seen in rejection. As well as causing a pneumonitis that has to be distinguished from rejection, cytomegalovirus may be involved in chronic lung rejection.122 It has been speculated that cytomegalovirus could promote allograft rejection by stimulating the production of proinflammatory cytokines or increasing the expression of major histocompatibility complex molecules.
The position of cytomegalovirus in regard to pulmonary disease in acquired immunodeficiency syndrome (AIDS) can also be difficult to determine. Cytomegalovirus inclusions are frequently encountered in AIDS but it is often difficult to determine whether pathological changes are due to the virus or to accompanying bacterial or Pneumocystis infection. Only occasionally is cytomegalovirus the only pathogen identified in severe pneumonia in AIDS patients.123 Some investigators claim that cytomegalovirus contributes to the high mortality from pneumonia in AIDS patients124 while others view it merely as a bystander rather than the primary pathogen in these patients.125 Sometimes, replication of the virus is unaccompanied by any significant degree of pulmonary inflammation or damage,126 indicating a poor host response, which, as in other viral infections, largely involves T lymphocytes, cells that are particularly defective in AIDS. The differing roles of cytomegalovirus in AIDS and transplant recipients have led to the view that the pathological changes are not a direct effect of the virus but an immunopathological condition attributable to the T-cell response to the virus.127
Pathological features
The pneumonia may be unilateral or bilateral, and generally involves the lower lobes; advanced lesions may appear as reddish purple nodular areas. Two patterns of pulmonary involvement have been described in bone marrow transplant recipients: a fulminant systemic infection characterised by a miliary pattern of disease and a more insidious disease with a more diffuse distribution in the lungs.121
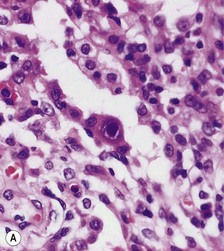
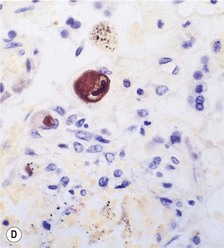
Histologically, there is a chronic interstitial pneumonitis and some of the alveolar epithelial cells are enlarged and contain characteristic inclusions. These measure up to as much as 10 µm in diameter and are surrounded by a clear zone inside the nuclear membrane (Fig. 5.1.13). These Cowdry type A intranuclear inclusions have been likened to an owl’s eyes. The inclusions represent clumped chromatin and the clear zone the virus. Cytoplasmic inclusions up to 2 µm in diameter are often also present. Severe cases may show a necrotising pneumonia or tracheobronchitis without the inclusions being well developed, in which case immunocytochemistry or in situ hybridisation may be used to advantage as these techniques show that many more cells are infected than those containing the characteristic inclusions (see Fig. 5.1.13D).6,7,128 Diffuse alveolar damage is a further pattern of disease that is occasionally seen in cytomegalovirus pneumonia.
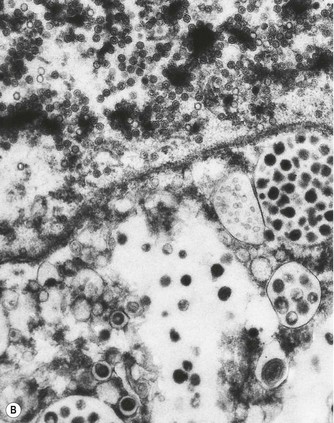
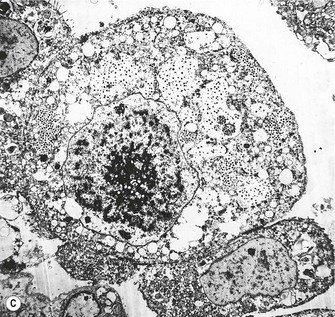
Figure 5.1.13 Cytomegalovirus pneumonia. (A) There is a prominent nuclear inclusion in the centre of the field. (B) Electron micrograph of an alveolar epithelial cell infected by cytomegalovirus. Numerous viral particles are evident in both nucleus (above) and cytoplasm (below). As the viral particles leave the nucleus and enter the cytoplasm they acquire a coating derived from the nuclear envelope and consequently enlarge. (C) Low-power electron micrograph of the cell seen in (B), showing that it is greatly enlarged compared with its neighbours. Coated viral particles are evident in the cytoplasm but uncoated particles in the nucleus are too small to be recognised at this magnification. However, characteristic central clumping of the chromatin is evident.
(B and C courtesy of Miss A Dewar, Brompton, UK.)
HIV and AIDS
Virology and means of spread
The HIVs belong to the lentivirus subfamily of the retroviruses and are thought to have originated from chimpanzees, which harbour the closely related simian immunodeficiency virus. They are the cause of AIDS and are transmitted primarily through sexual contact, by anal or vaginal intercourse with an HIV-positive person. Other routes of transmission are by exposure to infected blood, generally through the use of contaminated needles and syringes by drug addicts. Infected blood products and donor tissues are other potential sources of infection. An infected woman can pass the virus to her child in utero, at delivery or through breast-feeding. Occupational acquisition of HIV is unusual but has occurred, chiefly through needlestick injuries. In histopathology departments, particular care is required in handling unfixed tissues, as in preparing frozen sections and conducting autopsies. Fixed tissues do not present a risk of infection. Most national bodies have produced guidelines for safe laboratory practice in the AIDS era.129 HIV infection is not always recognisable and a practical approach is therefore to treat every cadaver and all unfixed tissue as if it were infectious.
Pathogenesis of AIDS
Following entry into the body and the development of neutralising antibodies, HIV is found in highest concentration within the germinal centres of lymphoid tissue where it can be demonstrated by immunohistochemistry attached to a follicular dendritic cell.129a The virus attacks both the follicular dendritic cells and the CD4+ helper T lymphocytes and blood levels of these latter cells below 200/µL are associated with the development of a variety of AIDS-defining conditions. The interferon-γ-secreting Th1 cells that are central to immune defence against a variety of other infections are particularly vulnerable to attack. Once HIV infection has occurred, antibody develops, generally within a month, and after a period that is usually measured in years CD4 counts drop and manifestations of AIDS develop. Concentrations of virus in the blood and body fluids are particularly high around the time of seroconversion and when AIDS develops.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


