Chapter 8 Vascular disease
8.1 Congestion and oedema; thrombosis, embolism and infarction; aneurysms
This section deals with several important pulmonary vascular diseases but some others are dealt with elsewhere – congenital anomalies of pulmonary vessels in Chapter 2, shock in Chapter 4 and vascular tumours of the lung in Chapter 12.3.
Pulmonary congestion and oedema
Congestion of the lungs may be active or passive, the former accompanying inflammation and the latter the result of an increase in pressure in the pulmonary veins, typically due to failure of the left ventricle or mitral stenosis. The active variety includes the congestive atelectasis that results from circulatory shock (see p. 143).
Pulmonary oedema
Pathogenesis
In accordance with Starling’s law, water moves between the vascular and interstitial compartments of the lung in response to net hydrostatic and osmotic forces acting across the vessel wall, modulated by the permeability of this membrane. The capillaries constitute the main site of microvascular fluid exchange, but small arteries and veins are also involved. Water movement chiefly takes place through the endothelial cell junctions; beyond these the endothelial basement membrane offers no barrier to fluid transport.1 Within the interstitium, water again flows along a hydrostatic gradient, in this case to the corners of the alveoli, and thence to the connective tissue surrounding the arteries and bronchioles at the centres of the acini and the veins in the interlobular septa.2,3 These are the sites where lymphatics commence.3,4 There is considerable reserve in the clearance capacity of the pulmonary lymphatics, which may increase their load 10-fold when pulmonary oedema threatens,5 but above this, spillover into the alveoli is inevitable. Before this the lungs sound dry on auscultation but the patient may nevertheless be breathless because the increased interstitial pressure triggers juxtacapillary nociceptors, which are represented by fine nerve terminals in the alveolar interstitium.6,7
In circumstances that involve only pressure changes, the oedema fluid is generally similar to normal interstitial fluid but in other circumstances the permeability of the vessels is increased and blood cells and high-molecular-weight plasma proteins, such as fibrinogen, escape into the tissues. Two main types of pulmonary oedema are therefore described, haemodynamic and cytotoxic (or irritant), characterised by the escape of low- and high-molecular-weight blood substances respectively. Electron microscopy shows that in haemodynamic oedema there is widening of the interstitial spaces in the alveolar walls by fluid, with separation of the collagen and elastin fibres, but that this is confined to the thick side of the air–blood barrier. In contrast, in cytotoxic oedema fluid also accumulates in the thin part of the air–blood barrier so that the endothelial cells are detached from their basement membranes and are often stretched over large subendothelial blebs of fluid (Fig. 8.1.1),8 changes that do not occur in uncomplicated haemodynamic pulmonary oedema.9
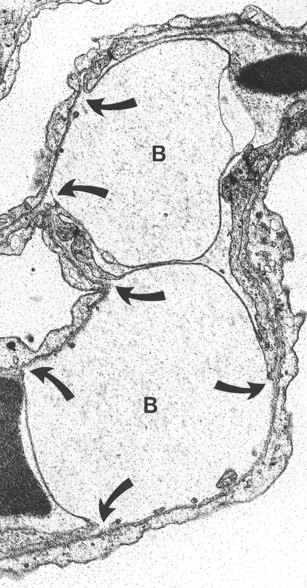
The separation of haemodynamic and cytotoxic oedema is not absolute: large haemodynamic forces alone can result in the escape of high-molecular-weight blood proteins.10,11 Electron microscopy shows that when the alveolar capillary pressure is raised above 40 mmHg in anaesthetised rabbits, breaks develop in both the endothelium of the alveolar capillaries and the alveolar epithelium.12–16 Such stress failure results in high-permeability pulmonary oedema, and even frank alveolar haemorrhage. This explains why the oedema fluid of mitral stenosis is often tinged with blood, and why haemosiderosis is seen in other diseases characterised by raised pulmonary venous pressure, such as veno-occlusive disease (see p. 427) and lymphangioleiomyomatosis (see p. 293).
Stress failure of the alveoli also explains the occasional sudden demise of a thoroughbred racehorse from a ‘burst blood vessel’.17 These animals have been bred to such an extent that their cardiovascular systems develop very high pulmonary vascular pressures on exercise. Alveolar haemorrhage is common in these horses, although it is seldom apparent to their attendants. Similar exercise-induced pulmonary haemorrhage has also been described in racing greyhounds and it is possible that the lungs of top athletes may also develop stress failure on occasion.15,18,19 Stress failure is also seen when there is overinflation of the lung, such as that which occurs when there is a sudden drop in atmospheric pressure (see ‘burst lung’, p. 369) or when alveolar gas pressures are raised unduly by mechanical respiratory assistance devices in intensive care units.
Specific causes of pulmonary oedema include:
Pathological findings
Microscopically, the interlobular septa and the periarterial connective tissue sheaths are swollen and lymphatics within them are dilated (see Fig. 2.8). Alveolar oedema is seen as a homogeneous, eosinophilic proteinaceous material filling the air spaces (Fig. 8.1.2). Entrapped air bubbles are represented by rounded spaces within the proteinaceous contents of the alveoli. Only scanty threads of fibrin are generally found, but in severe cases, especially of the toxic variety, much fibrin is present in the fluid. The lungs are then particularly firm and have been described as showing ‘congestive consolidation’. There may also be many red blood cells in the air spaces. Iron-containing macrophages may be found in chronic cases.43a
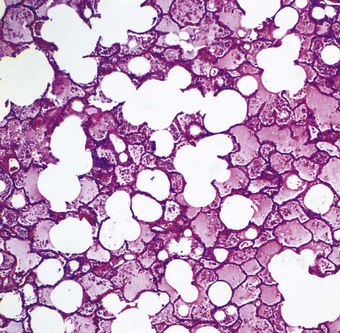
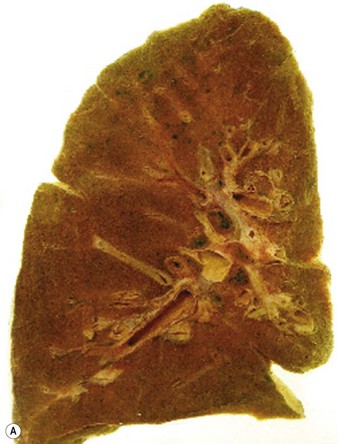
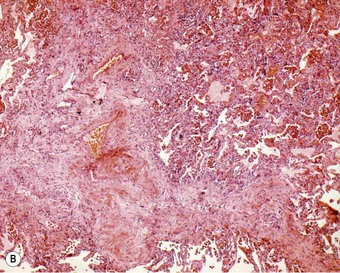
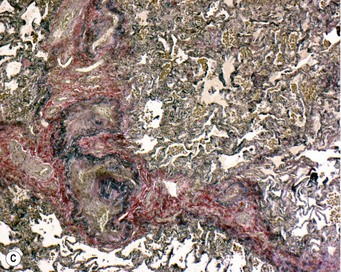
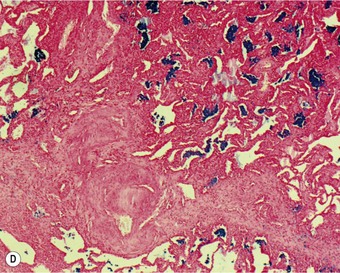
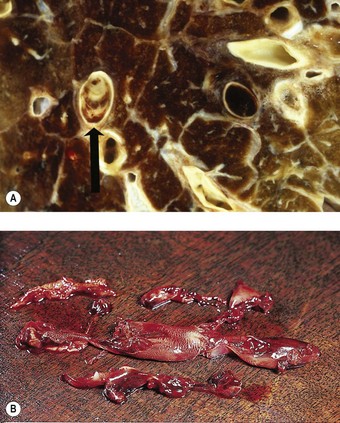
Organisation of the fibrinous material may occur, leading to fibrosis in the alveoli.44 This takes the form of intra-alveolar plugs of oedematous granulation tissue known as bourgeons conjonctifs or Masson bodies.45 Similar bodies develop in the bronchioles and the appearances closely mimic those found in healing bacterial pneumonia and in cryptogenic organising pneumonia. The connective tissue plugs may be incorporated into the alveolar walls, causing interstitial fibrosis by an accretive process.46,47 Interstitial fibrosis may also arise as a direct consequence of interstitial oedema, so-called gefässlose (vessel-less) organisation.48 The combination of haemosiderosis and fibrosis spawned the term ‘brown induration of the lungs’ for the changes that develop in long-standing mitral stenosis (Fig. 8.1.3).49
Figure 8.1.3 ‘Brown induration’ representing a combination of haemosiderosis and interstitial fibrosis due to chronic pulmonary congestion and oedema in patients with long-standing left-sided cardiac failure. (A) Paper-mounted whole-lung section showing brown discoloration.
(Courtesy of WG Edwards, London, UK.)
The vascular changes resulting from pulmonary venous hypertension are described in Chapter 8.2.
Radiological findings44,50,51
Interstitial oedema is recognisable in radiographs of the chest by a combination of hilar enlargement, caused by peribronchial and periarterial cuffing, and peripheral linear opacities resulting from oedema of the interlobular septa. There is a greater concentration of perivascular and peribronchial connective tissue at the hila and consequently the hilar opacities are enlarged, more dense and less well defined. Septal thickening is visualised radiologically as a series of non-branching, horizontal lines, 1–3 cm long, extending to the pleura at the lung bases. They are popularly known as Kerley B lines.
In addition to the features of oedema, the chest radiograph often shows enlargement of the heart and widening of vessels to upper zones of the lungs. The latter may be a direct effect of raised venous pressure on vessels that are normally at lower than alveolar pressure and therefore collapsed, or it may be a consequence of the vascular remodelling seen at the lung bases in pulmonary venous hypertension, which is described on page 427.
Clinical features
Breathlessness is an early feature of pulmonary oedema, encountered when the process is still interstitial because of stimulation of the juxtacapillary nociceptors.6,7 With the onset of alveolar oedema crepitations are heard on auscultation and in severe cases watery sputum is produced, sometimes tinged by blood, and the patient is cyanosed and hypoxaemic. It is often essential to maintain cerebral oxygenation by augmenting the concentration of inspired oxygen despite the deleterious effect this may have on an already injured lung. Patients in left ventricular failure are prone to attacks of severe breathlessness when recumbent, typically at night. This is aptly termed paroxysmal nocturnal dyspnoea but is often still referred to by the older term ‘cardiac asthma’. The distinction from bronchial asthma (see p. 109) may be blurred by oedematous swelling of the bronchial wall causing the patient to wheeze.
Pulmonary thrombosis and thromboembolism
Pulmonary thrombosis, as distinct from thromboembolism, is seldom symptomatic in the absence of other pulmonary vascular disease. It is most commonly encountered as a complication of pulmonary hypertension of any cause, and then confounds the histological distinction of thromboembolic pulmonary hypertension from that due to other causes, in particular the primary variety. It is necessary therefore to search for the specific histological features of the other varieties of pulmonary hypertension (which are dealt with in the next chapter) whenever pulmonary thrombosis is recognised. Although atheroma is a feature of pulmonary hypertension (see Fig. 8.2.17),52 it is generally confined to the large elastic pulmonary arteries and seldom attains the advanced, ulcerated degree that predisposes to aortic thrombosis. Thrombosis complicating pulmonary hypertension probably commences in the smaller muscular pulmonary arteries, which narrow in this condition.
As well as complicating pulmonary hypertension, pulmonary thrombosis and infarction also develop in systemic lupus erythematosus,53 sickle cell disease,54–58 spherocytosis,59 thalassaemia,60,61 paroxysmal nocturnal haemoglobinuria,62 the antiphospholipid antibody syndrome,53,63 the hypereosinophilic syndrome,64,65 rare varieties of pulmonary vasculitis that affect the large elastic arteries,66,67 diffuse alveolar damage and unusual varieties of vascular infection.68 Pulmonary thrombosis is also predisposed to by low blood flow, as in congenital anomalies such as pulmonary stenosis and in pulmonary aneurysms.
Thromboembolism
Aetiology
Pulmonary thromboembolism is rare in the third world but common in developed countries.69 Economic development in Hong Kong has been accompanied by an increasing incidence of the disease70,71 and in the USA the incidence of pulmonary thromboembolism among non-whites is much the same as in whites.72
In western countries, thrombotic pulmonary emboli, often unsuspected in life, are commonly encountered at necropsy, especially in elderly and obese people who have been confined to bed with congestive cardiac failure, and in adults of any age who have undergone major surgery, suffer from cancer or have been the victims of major trauma.73–79 They develop in people who sleep in chairs that hinder venous return from the legs (e.g. deck chairs) and especially in air-travellers who are immobile for a long time. Nearly half prove fatal.80 Pulmonary embolism is also prone to occur after childbirth. Contributing factors are listed in Box 8.1.1.81–83 They operate in three ways, as described by Virchow in his well-known triad:
Box 8.1.1 Risk factors for venous thromboembolism83
aInflammatory bowel disease, nephrotic syndrome, chronic dialysis, myeloproliferative disorders, paroxysmal nocturnal haemoglobinuria, Behçet’s disease.
Thrombosis after operations and childbirth is promoted by the increased numbers of platelets that accompany any form of trauma. It often occurs in tributaries of the main veins of the legs and thighs, where the vascular stasis that follows immobilisation is known to be most pronounced. Puerperal thromboembolism is more frequent in women whose lactation has been suppressed by the administration of oestrogens than among those who breast-feed and in line with this it is known that the older high-oestrogen contraceptive pill carried a small risk of promoting thromboembolism by affecting the clotting factors VII and X and platelet aggregation. This risk is believed to be almost non-existent with the present low-oestrogen contraceptive pills. 84
Hypercoaguability is fairly prevalent in the general population and presumably augments the other factors contributing to thrombosis listed in Box 8.1.1. It can be recognised in 25% of patients with venous thromboembolism and in an even higher number of patients whose thrombosis is otherwise unexplained. High levels of factors VIII and XI are fairly common in the general population (each has a prevalence of about 10%) but abnormalities in the levels of many other clotting factors are also recognised.85 They include deficiencies of antithrombin III, factor V (Leiden), protein C and protein S. Usually these need to interact with acquired risk factors before thrombosis occurs, being otherwise uncommonly associated with idiopathic thromboembolism. For example, factor V (Leiden) defect in isolation increases the risk of thromboembolism 3–5-fold but in conjunction with oestrogen therapy 35-fold.83 Further conditions characterised by hypercoaguability include the antiphospholipid syndrome and microalbuminuria.63,86
Despite the dangers of childbirth and oral contraception, the standardised mortality rate from thromboembolism is roughly the same for men and women; the increased risk of major trauma in young male adults balances the risks special to women. However, the crude death rate from thromboembolism is much greater in women than in men87 because of the marked effect of advancing age on the incidence of thromboembolism and the greater number of elderly women in the population (Table 8.1.1). There is also a minor seasonal variation in the incidence of pulmonary thromboembolism, the risk being greater in the colder months (Table 8.1.2).88–92
Table 8.1.1 Deaths from pulmonary embolism per million population by age and sex in England and Wales

Table 8.1.2 Deaths from pulmonary embolism by seasona
| Season | Deaths (n) |
|---|---|
| January to March | 2898 |
| April to June | 2651 |
| July to September | 2646 |
| October to December | 2893 |
a 11088 cases assembled from 23 reports from the northern hemisphere.90
It is when calf vein thrombi propagate proximally to reach the main veins of the thigh that they are most likely to break off and reach the lungs as emboli (Table 8.1.3).93 Proximal propagation is encountered in approximately 20% of patients with calf vein thrombosis and approximately 50% of those with proximal leg vein thrombosis develop pulmonary embolism.94 Residual deep-vein thrombosis can be demonstrated in most patients who have recently suffered pulmonary embolism.95 The danger of leg vein thrombosis and embolism is lessened by the use of prophylactic massage and exercise. Treatment is mainly based on anticoagulation but may involve venous ligation proximal to the thrombus and thromboembolectomy. Leg vein thrombosis is nearly always bilateral and this should be kept in mind when ligation is being considered. Approximately 11% of patients with pulmonary thromboembolism die within 1 hour and only 29% of the remainder are correctly diagnosed and treated appropriately, which is unfortunate as there are 10 times as many deaths in the undiagnosed and untreated majority (Fig. 8.1.4).74
Table 8.1.3 Incidence of pulmonary embolism according to the site of leg vein thrombosis93
| Pulmonary embolism | Incidence |
|---|---|
| Calf vein thrombosis | 0/21 patients |
| Calf and thigh vein thrombosis | 8/15 patients |
Pulmonary embolism detected by lung perfusion and ventilation scans.
Leg vein thrombosis detected by venography, impedance plethysmography and radiofibrinogen scanning.
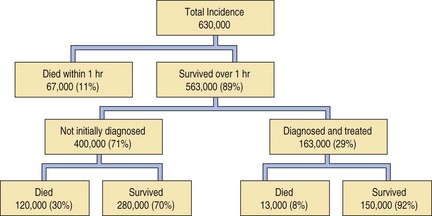
Figure 8.1.4 Annual incidence and survival in pulmonary thromboembolism in the USA.
(Reproduced from Dalen & Alpert (1975)74 with permission.)
The periprostatic venous plexus is another source of pulmonary emboli, particularly multiple small emboli. Thrombosis here is promoted by local inflammation as well as by sluggish venous flow as in heart failure. Less often, thrombotic emboli originate in the veins of the arms or in the chambers on the right side of the heart. Other sources include intravenous cannulae and endocarditis affecting the valves on the right side of the heart. In one study 86% of thrombotic emboli reached the lungs via the inferior vena cava, 3% came via the superior vena cava, 3% originated in the heart and in 8% of cases the thrombosis was widespread.96
Clinical features
Sometimes the embolus is a massive one, and its lodgement in the pulmonary trunk or main pulmonary artery is immediately fatal because it obstructs the pulmonary circulation. In only 28% of such cases is a diagnosis of pulmonary thromboembolism made before death.96 More frequently the embolus is small and without significant effect. Alternatively, the embolus may result in pulmonary infarction, which is indicated clinically by an attack of localised pleural pain, dyspnoea and haemoptysis. Often there is more than one embolic episode. Multiple microemboli as a cause of pulmonary hypertension is considered on page 426.
The high prevalence of unsuspected pulmonary thromboembolism evident at autopsy has been demonstrated in life with perfusion scans, emphasising that once a diagnosis of peripheral venous thrombosis has been established, anticoagulant therapy should be instituted without delay.97 Thromboendartectomy is being increasingly undertaken.98,99
Distribution of the emboli
When clots are introduced into a rabbit’s systemic vein, both the size of the clots and the anatomy of the pulmonary arterial tree affect their subsequent distribution in the lungs.100 Whilst large emboli lodge in any of the main arteries, those of medium size are carried disproportionately often into the vessels of the posterior basal segments. In humans, pulmonary infarcts are commoner in the posterior basal segments of the lower lobes96 and solitary metastatic tumours are more frequently basal than apical. The explanation lies in the anatomy of the pulmonary arteries: they have a large axial trunk that gives off its branches at an angle and terminates in the posterior basal segment.
Pathological findings
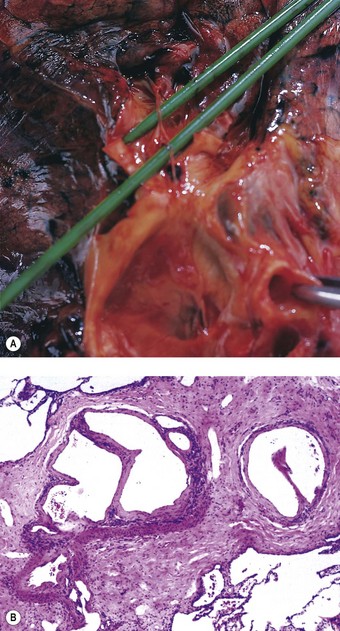
The sudden impaction of a large embolus in the pulmonary trunk brings the circulation virtually to a halt. The small volume of blood that can percolate past the thrombotic mass is quite insufficient to fill the left side of the heart and maintain normal systemic arterial pressure and an adequate circulation through the cerebral and coronary arteries. In such cases, the embolus is found at necropsy to be coiled on itself, straddling the bifurcation of the pulmonary trunk (Fig. 8.1.5). The chambers of the right side of the heart and the venae cavae are distended, the left atrium and ventricle contracted, and the lungs pale. The coiling distinguishes embolic thrombi from thrombi that have formed in the pulmonary arteries. When the embolus is removed from its site of impaction and is uncoiled it is seen to be of a diameter that corresponds to the calibre of the vessels in which it formed. Its surface is marked in correspondence with the venous valves: the broken ends of the thrombotic cast of the tributary veins can also be recognised and it may be possible to match these with the thrombus remaining in the veins.
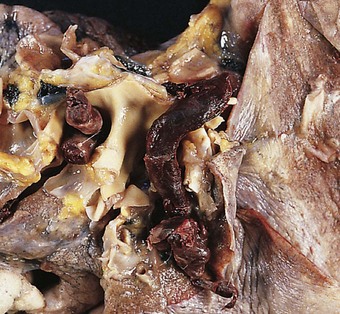
Figure 8.1.5 Massive pulmonary thromboembolism. Coiled thrombus occludes the pulmonary trunk and the main pulmonary arteries.
(Courtesy of Dr GA Russell, Tunbridge Wells, UK.)
Both thrombi formed in situ and thrombotic emboli are firm but friable, and show striae of Zahn, which are evident to the naked eye and indicative of their development during life (Fig. 8.1.6): these features distinguish them from clots, which are formed postmortem. Clots are shiny, soft and elastic: when pulled carefully from the vessels they show a characteristic ‘horse’s tail’ appearance, their end being a cast of blood from the smaller branches of the pulmonary artery. Clots may show a transition from red to yellow, due to postmortem sedimentation of the cellular constituents of the stagnant blood, leaving a paler zone of serum in the eventual coagulum: this demarcation lacks the alternate white and red layers formed respectively of platelets and fibrin-enmeshed blood cells that characterise the striae of Zahn in a thrombus.
Fresh thrombus may fragment and disperse into smaller pulmonary arteries, of which there is a great reserve, but within a few days thrombotic emboli undergo organisation and become firmly adherent to the vessel wall.98,99 In 4–6 weeks they are converted into fibrous tissue, often with recanalisation. Some emboli seem to disappear and it is presumed that they are destroyed by fibrinolysin. Thin fibrous bands stretching across the lumen of major pulmonary arteries are sometimes the only evidence of previous thromboembolism (Fig. 8.1.7A).101 More often, the lumen of smaller arteries is divided up into multiple channels by the usual process of recanalisation (Fig. 8.1.7B).102
Pulmonary infarction
Like other infarcts, those in the lungs are generally wedge-shaped. An occluded pulmonary artery is found at the apex of the infarct; the base of the infarct is on the pleura. Often the edge of a lobe is involved, in which case the lesion is diamond-shaped rather than wedge-shaped (Fig. 8.1.8). Any part of the lung may be affected but infarction is commonest at the bases, for the reasons explained above. Pulmonary infarcts are commonly multiple.

Partial and complete infarction
Partial infarcts are initially characterised by oedema but within 48 hours capillaries rupture and the resultant lesions are intensely haemorrhagic, swollen, firm and dark red (see Fig. 8.1.8). The edge is sharply demarcated. Acute inflammation develops where infarcts border healthy lung and fibrin is found on the pleural surface of the involved area. Microscopically, the alveoli are filled with blood. Their walls are intact however and the basic architecture of the tissue is unaltered. Complete and rapid resolution is frequently observed radiologically and if the patient then dies, perhaps of a further massive embolism, no abnormality other than haemosiderin-laden macrophages can be detected in the recently infarcted area. Less often, necrosis is found, signifying true infarction. Healing is then by organisation, which is marked by slow paling and shrinkage, until eventually only a barely detectable scar remains. As with most scars in organs that are subject to constant movement, such as the lung and the heart, they are rich in elastin.103 If the lesion is excised before it has been fully organised, the central necrosis is seen to be bordered by granulation tissue rich in foam cells and haemosiderin-laden macrophages. However, if these features are not well developed conditions such as Wegener’s granulomatosis, rheumatoid nodule and chronic infection enter the differential diagnosis, especially if the classic wedge shape of an infarction is not well represented.104 Basophilic karyorrhexis and chronic inflammation involving both the granulation tissue and the wall of the artery in which the embolus impacted may further the impression of Wegener’s while palisading of the reparative cells may suggest a rheumatoid nodule. In these circumstances the diagnosis of infarction is often no more than tentative, based partly upon negative features, such as an absence of extrapulmonary and serological evidence of Wegener’s granulomatosis and rheumatoid disease and no evidence of infection being found.
Pulmonary venous infarction
In contrast to the infarcts caused by pulmonary artery occlusion, pulmonary venous infarction is very rare. This is attributable to the rich network of venous collateral vessels that drain the lung. Most of the few reported cases have been due to sclerosing mediastinitis. Other recorded causes include left atrial myxoma, left atrial thrombosis complicating mitral stenosis, venous thrombosis following pulmonary resection and carcinoma of the lung.105,106 The infarcts are similar to those caused by arterial occlusion. Chronic venous obstruction, as seen in pulmonary veno-occlusive disease (see p. 427) may result in interstitial fibrosis. These areas of scarring are rich in haemosiderin and possibly represent healed venous infarcts.
Non-thrombotic pulmonary emboli
Pulmonary embolism may result from various kinds of material forming in, or gaining access to, the systemic veins. Apart from thrombi, these include fat droplets, various types of tissue, clumps of tumour cells, gas bubbles and a variety of foreign substances. In some parts of the world parasitic emboli are important: these include the eggs of schistosomes (p. 254) and the adult form of the canine heart worm Dirofilaria immitis (p. 258).107 In contrast to thrombotic emboli, the effects of non-thrombotic emboli are not purely mechanical but are also linked to the nature of the embolus.
Fat embolism
Fat embolism is usually the result of bone or soft-tissue trauma, but other causes such as liposuction or bone marrow infarction complicating haemoglobinopathy are occasionally responsible (Box 8.1.2).107–111 Under any of these circumstances, fat or oil globules enter the systemic veins and reach the lungs. Small fat globules can almost always be demonstrated in pulmonary capillaries after any fracture if frozen sections are stained appropriately and only a small proportion of patients develop symptoms referable to the embolism.
Some patients with fat embolism develop severe respiratory failure. This is seldom due to extensive occlusion of the pulmonary circulation. More often it is caused by traumatic shock, which has its own profound effects on the lungs (see p. 142). Alternatively, it is suggested that the lungs are damaged by irritant fatty acids released by lysis of the fat.112–114
Microembolism of agglutinated fat emulsions110,115,116 or of crystals derived from infusions during the course of total parenteral nutrition117 is described on page 390.
Amniotic fluid embolism
Occasionally, during childbirth, amniotic fluid with its content of particulate debris, mostly epidermal squames and meconium, is forced by strong uterine contractions into the maternal circulation through small incomplete lower uterine tears, resulting in fatal microembolism of the mother’s small pulmonary arteries.118 The emboli may be quite sparse, suggesting that death is due to anaphylactic shock rather than pulmonary vascular occlusion, a possibility that is supported in some cases by the amniotic fluid embolism being accompanied by disseminated intravascular coagulation (see p. 144), mast cell degranulation and complement activation.119 Recognition of the meconium mucus and epidermal squames is facilitated by the application of a mucous stain such as alcian blue and immunohistochemistry using antibodies against cytokeratin (coupled with an endothelial marker such as CD34 to distinguish the squames from degenerate exfoliated endothelial cells).
Trophoblastic embolism
Trophoblastic tissue may be found in the lungs in a large proportion of women who die during pregnancy or the puerperium.120 It is particularly common in women suffering from eclampsia and in those undergoing uterine surgery for hydatidiform moles and as a side-effect of chemotherapy for choriocarcinoma.121 Trophoblastic embolism is recognisable as isolated multinucleated syncytiotrophoblastic cells within pulmonary blood vessels (Fig. 8.1.9) and can be confirmed as such by the immunocytochemical demonstration of human chorionic gonadotrophin. Chorionic villi are not observed in the pulmonary vessels in normal pregnancies but are common in molar pregnancies.122 It is believed that uterine contractions dislodge the trophoblastic tissue and enable it to enter the blood stream. The trophoblastic emboli cause no structural changes in the lung and appear to be immunologically inert, in spite of their fetal origin. They do not predispose to thrombosis and usually undergo early lysis120 but rare fatal cases of massive trophoblastic embolism are recorded in which over 80% of the pulmonary vessels are occluded.123 In exceptional cases of benign hydatidiform mole, it has been noted radiologically that masses within the lungs may persist for up to a year, but regress and totally disappear thereafter124; they presumably represent trophoblastic emboli that have gained a temporary foothold in the lungs.
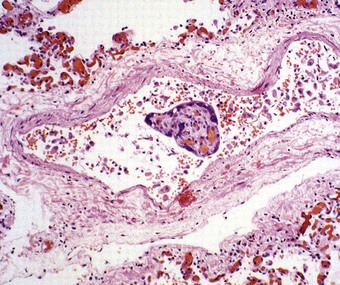
Decidual embolism
Small foci of decidua are occasionally observed in the lungs in pregnancy. Such foci located immediately beneath the pleura125 may represent focal metaplasia of the overlying serosa but deep in the lung an embolic aetiology is generally assumed, although the decidual tissue is usually extravascular. The nature of this tissue is discussed further under the subject of pleuropulmonary endometriosis (see p. 495).
Tumour embolism
Tumour emboli are common in the lungs. A few survive to form metastatic deposits but most die very soon after arriving in the lungs: the embolus then becomes enclosed by platelets and finally undergoes organisation.126 Vascular occlusion by tumour emboli is sometimes so widespread that it leads to right-sided heart failure127–133 whilst massive tumour emboli have sometimes proved fatal134–136 or necessitated emergency embolectomy.137 In other patients they have caused flitting radiographic opacities, which presumably represent infarcts.138 Occlusion of the pulmonary arteries may be due to tumour alone or the tumour may promote thrombosis, organisation of which results in fibrocellular intimal thickening, so-called carcinomatous thrombotic microangiopathy or vascular intimal carcinomatosis (see p. 682).139–142 Embolic sarcoma is distinguished from the primary sarcoma of the pulmonary arteries described on page 638 by the presence of an extrapulmonary source.
Other tissue embolism
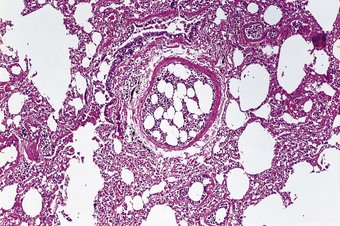
Bone marrow is frequently seen plugging small pulmonary arteries postmortem when ribs have been broken during unsuccessful attempts at cardiac resuscitation or following trauma to other bones (Fig. 8.1.10). Other traumatic emboli may consist of brain tissue,143,144 liver tissue,145,145a bile (Fig. 8.1.11),146 bone fragments (Fig. 8.1.12) or very rarely atheromatous cholesterol crystals.147 Haemorrhage has been ascribed to the last of these but none of the others is of clinical consequence; they merely represent incidental necropsy findings.
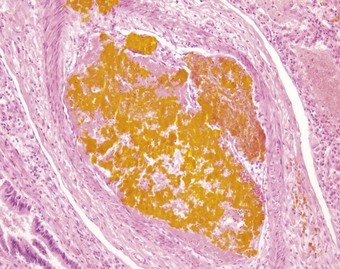
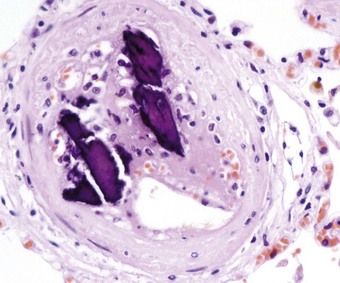
Foreign-body embolism
Some drug addicts break up preparations of drugs intended for oral use and inject an aqueous suspension intravenously. Substances used as fillers in the manufacture of tablets include starch, cellulose, talc and crosprovidone (poly[N-vinyl-2-pyrrolidone]). When injected intravenously, these lodge in pulmonary vessels and induce thrombosis and a local foreign-body granulomatous reaction (Fig. 8.1.13 and see Fig. 7.2.10, p. 379).148–155 Pulmonary hypertension may result if the injury to the vessels is extensive.148 Widespread pulmonary fibrosis may also develop, occasionally mimicking pneumoconiotic progressive massive fibrosis.156 Starch particles can be identified by their characteristic Maltese cross appearance on polarising microscopy, and talc by its platy form on polarising microscopy and by elemental electron microprobe analysis.156 Cellulose fibres can be distinguish from platy crystals such as talc by their reaction with periodic acid–Schiff, silver methenamine and Congo red stains.153,157 Crosprovidone is seen as basophilic, coral-like particles, the nature of which can be confirmed by infrared spectroscopy.158 Any of these particles may leave the blood vessels and be found in the interstitial tissues and even air spaces. Talc particles in the lung exceeding 5 µm in length should arouse suspicion of intravenous drug abuse,159 but cellulose fibres up to 120 µm in length may reach the lungs and cause granulomatosis in addicts who inhale their illicit drugs.157,160
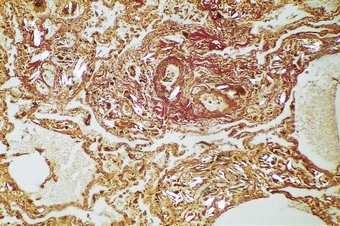
Other embolic foreign bodies described in pulmonary arteries include airgun pellets,161 shrapnel, whole bullets,162 masonry and stone fragments, food particles (Fig. 8.1.14), the broken-off ends of intravenous catheters and cannulas, plastic particles from dialysis tubing and other intravascular devices,163,163a therapeutic injections or prosthetic implants of substances such as Teflon, hyaluronic acid and silicone,164a,168a lymphangiography contrast medium,169–172 various materials injected to occlude abnormal blood vessels or deprive a tumour of its blood supply,173,174 and mercury, this last generally introduced into the circulation with suicidal intent.175 Patients with psychiatric problems have also injected themselves with various other substances, such as olive oil, resulting in pulmonary lipogranulomatosis.176
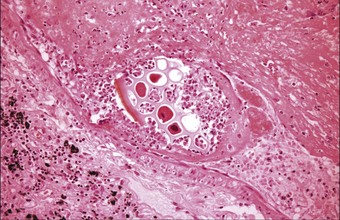
Figure 8.1.14 Embolised vegetable matter is present within a pulmonary artery in a patient who died of a ruptured oesophagus.
(Courtesy of Dr CA Seldenrijk, Nieuwegein, The Netherlands.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


