Cystic Fibrosis
KEY CONCEPTS
![]() Good nutrition with appropriate pancreatic enzyme and vitamin supplementation are essential in the management of cystic fibrosis (CF).
Good nutrition with appropriate pancreatic enzyme and vitamin supplementation are essential in the management of cystic fibrosis (CF).
![]() Airway clearance and antiinflammatory therapies are key components to improve pulmonary health in CF patients.
Airway clearance and antiinflammatory therapies are key components to improve pulmonary health in CF patients.
![]() Antipseudomonal agents are the cornerstone of antibiotic therapy for chronic lung infections in the CF patient.
Antipseudomonal agents are the cornerstone of antibiotic therapy for chronic lung infections in the CF patient.
![]() Altered pharmacokinetics of CF patients can impact the dosing and clearance of pharmacologic therapy.
Altered pharmacokinetics of CF patients can impact the dosing and clearance of pharmacologic therapy.
“Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon must die.” This European adage accurately describes the fate of an individual diagnosed with cystic fibrosis during ancient times.1
Cystic fibrosis (CF) is a disease state resulting from a dysfunction in the cystic fibrosis transmembrane conductance regulator (CFTR). It is the most common life-limiting disorder in the Caucasian population, with an incidence of 1 in 2000 to 4000 live births and a prevalence of 30,000 affected individuals in the United States.2–7
Currently with care, affected individuals have an expected life span of 36 years of age. Multiple organ systems are affected in CF individuals, especially, the lungs, the digestive system, and the reproductive organs. Mortality is most commonly due to chronic organ damage or resistant pulmonary infections.8
The pharmacist plays an essential role in the development and management of a pharmacotherapeutic care plan for the CF patient.
EPIDEMIOLOGY
Cystic fibrosis occurs in approximately 1 in 3,500 newborns. In the 1970s, patients only survived into their teen years. By 2006, progress in care had extended survival to 36 years. Institution of care at a young age impacts long-term survival; hence, timing of diagnosis and recognition of signs and symptoms are crucial.2–7
Although CF occurs in all ethnicities, other ethnicities besides the Caucasian population display lower frequencies: 1 in 9,200 Hispanic-Americans, 1 in 10,900 Native Americans, 1 in 15,000 African Americans, and 1 in 100,000 Asians. The carrier frequency is 1 in 28 North American white populations, 1 in 29 Ashkenazi Jews, and 1 in 60 African Americans.2
ETIOLOGY
The cause of CF is due to a mutation of the CFTR gene. Extensive genetic studies have increased awareness regarding the large spectrum of mutations in the CF population. Over 1,800 mutations have been identified due to the extensive collaboration of the CF Foundation with international researchers. The most common mutation identified in CF patients is ΔF508.3
CF is an autosomal recessive disease, in which one mutation present on each allele of the CFTR gene results in presentation of the disease. The presentation of a mutation on only one allele of the CFTR gene will prevent the full expression of CF. Genetic studies have increased the understanding of genotype–phenotype relationships. Various mutations on the CFTR gene can result in various pathologies such as primary lung disease to minor GI involvement.3
PATHOPHYSIOLOGY
In order to successfully treat CF, a good understanding of the disease’s underlying mechanism of action is crucial. It is well established that gene mutations cause an abnormality in the cystic fibrosis transmembrane conductance regulator (CFTR). This initiates the sequence of events responsible for the manifestations of CF. Mucosal obstruction occurs in the distal airways of the lung and submucosal glands, which express the CFTR. The CFTR also performs numerous cellular functions, including the regulation of chloride transport across the cell membrane. Studies in genotype–phenotype relationships have shown that an abnormality on the CFTR contributes to the expression of other gene proteins involved with inflammatory responses, ion transport, and cell signaling. These various expressions result in differences in clinical severity among patients with the same mutations on the CFTR.3,9–11
Under normal conditions, the CFTR helps regulate ion transport and salt homeostasis in the sweat glands. Typically, the sodium ion is followed by the chloride ion, and is reabsorbed from the lumen by the CFTR and apical sodium channels. As a result of the CFTR’s malfunction in CF patients, chloride fails to be reabsorbed, which impacts the sodium ion reabsorption as well. This failed process produces sweat that contains high levels of salt. The endpoint of this process is a highly negatively charged lumen, which leads to an increased salt content in the sweat gland. This is known as the transepithelial potential difference, which is two to three times greater in CF patients. These processes can lead to organ damage in the CF patient (Fig. 18-1).
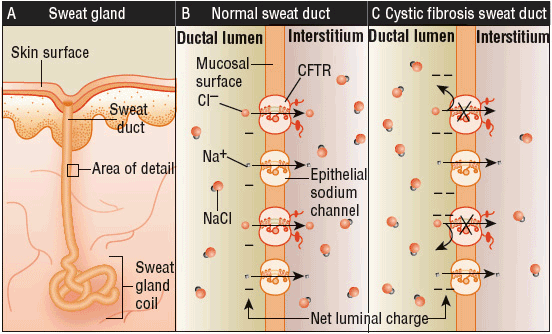
FIGURE 18-1 Mechanism of underlying elevated sodium chloride levels in the sweat of patients with cystic fibrosis (CF). Sweat ducts (Panel A) in patients with CF differ from those in people without the disease in the ability to reabsorb chloride before the emergence of sweat on the surface of the skin. A major pathway for Cl– absorption is through CFTR, situated within luminal plasma membranes of cells lining the duct (i.e., on the apical, or mucosal, cell surface) (Panel B). Diminished chloride reabsorption in the setting of continued sodium uptake leads to an elevated transepithelial potential difference across the wall of the sweat duct, and the lumen becomes more negatively charged because of a failure to reabsorb chloride (Panel C). The result is that total sodium chloride flux is markedly decreased, leading to increased salt content. The thickness of the arrows corresponds to the degree of movement of ions. (From Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005;352(19):1992–2001.)
There are several theories as to how mucosal obstruction occurs in the airways. One of these theories, known as the “low-volume model,” explains that the pulmonary surface epithelium behaves the opposite of a sweat gland in CF patients. There is an increased absorption of sodium, chloride, and fluid, which causes dehydration of the airway surfaces and defective mucociliary transport. An alternative theory known as the “high-salt model” indicates that the pulmonary surface epithelium behaves similarly to the sweat gland. A high salt content predisposes the CF patient to bacterial infections. Both theories agree that CF airways lack the ability to transport chloride through the CFTR.9,12,13
One of the common causes of morbidity and mortality in CF patients is mucosal obstruction of the exocrine glands. Mucosal obstruction causes the ducts to dilate, which results in the coating of lung surfaces by thick, viscous, neutrophil-dominated debris. These secretions initiate a cascade of events that lead to inflammation and formation of scar tissue in the lungs14 (Fig. 18-2).
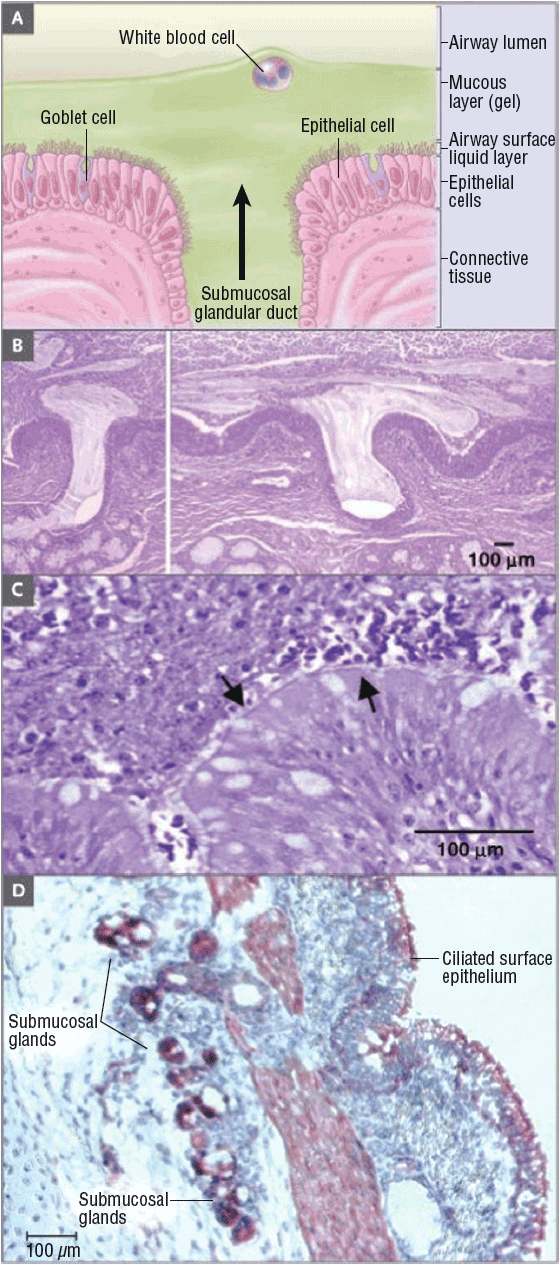
FIGURE 18-2 Extrusion of mucus secretion onto the epithelial surface of airways in cystic fibrosis. Panel A shows a schematic of the surface epithelium and supporting glandular structure of the human airway. In Panel B, the submucosal glands of a patient with CF are filled with mucus, and mucopurulent debris overlies the airway surfaces, essentially burying the epithelium. Panel C is a higher-magnification view of a mucus plug tightly adhering to the airway surface, with arrows indicating the interface between infected and inflamed secretions and the underlying epithelium to which the secretions adhere. (Both Panels B and C were stained with hematoxylin and eosin, with the colors modified to highlight structures.) Infected secretions obstruct airways and, over time, dramatically disrupt the normal architecture of the lung. In Panel D, CFTR is expressed in surface epithelium and serous cells at the base of submucosal glands in a porcine lung sample, as shown by the dark staining, signifying binding by CFTR antibodies to epithelial structures (aminoethylcarbazole detection of horseradish peroxidase with hematoxylin counterstain). (From Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005;352(19):1992–2001.)
Other organ systems are impacted by the absence of CFTR activity as well. Ten percent of CF patients are born with meconium ileus, which is an intestinal obstruction that may be fatal if left untreated. Blockage of the pancreatic duct leads to complications such as chronic fibrosis and fatty replacement of the pancreatic gland. Bile duct obstruction causes cirrhosis of the liver, and male CF patients can experience infertility due to obstruction of the vas deferens in utero.9
CLINICAL PRESENTATION
TABLE 18-1 Cystic Fibrosis Foundation Diagnosis Criteria and Clinical Presentation
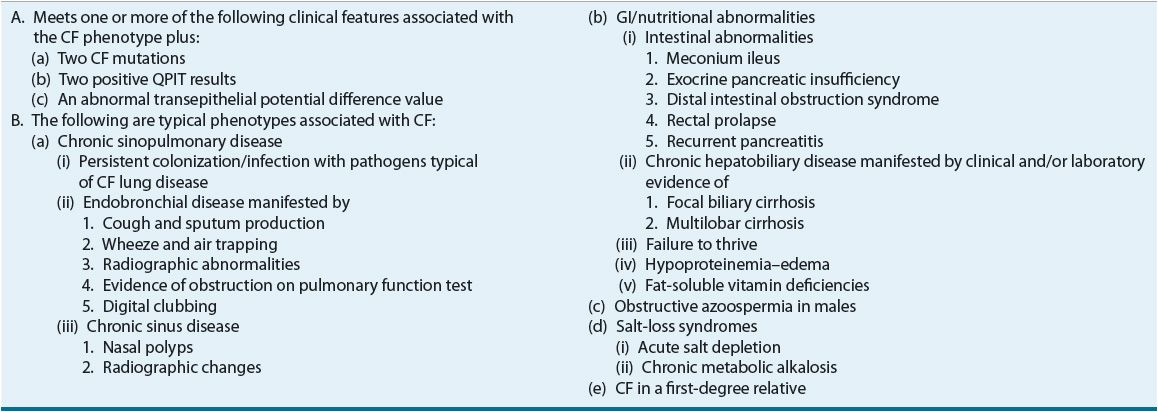
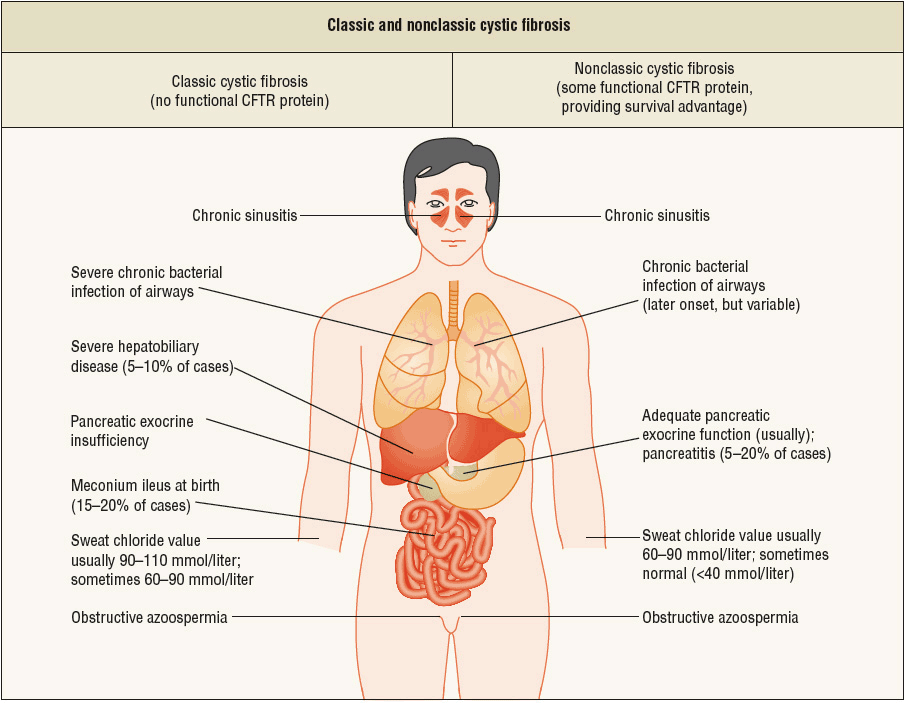
FIGURE 18-3 Classic and nonclassic cystic fibrosis (CF). The findings in classic CF are shown on the left-hand side, and those of nonclassic CF on the right-hand side. Patients with nonclassic CF have better nutritional status and better overall survival. Although the lung disease is variable, patients with nonclassic CF usually have late-onset or more slowly progressive lung disease. Sweat-gland function, as evidenced by the sweat chloride test, is abnormal but not to the extent noted in classic CF. Pancreatitis may occur in patients with nonclassic disease. However, chronic sinusitis and obstructive azoospermia occur in both groups of patients. On the basis of these findings, one can infer that mutations in CTFR, perhaps coupled with other genetic or environmental factors, may confer a predisposition to sinusitis, pancreatitis, or congenital bilateral absence of the vas deferens (azoospermia) in the general population. (From Knowles MR, Durie PR. What is cystic fibrosis? N Engl J Med 2002;347(6):439–442.)
Sinus and Pulmonary Presentation
Cystic fibrosis patients will usually experience chronic infections and frequently develop polyps in the sinus cavity. Daily symptomatology includes shortness of breath and cough, with sputum production. A common finding in radiology chest films is a flat diaphragm with an increased chest diameter and air trapping. Pulmonary function tests will reflect a decrease in FEV1. Older patients will experience digital clubbing, a deformity of the fingers and fingernails often associated with chronic hypoxia.
Bacterial growth in the lungs will often drive CF patients to a state of exacerbation, resulting in increased cough, a reduction in pulmonary function, and increased sputum production with a change in color.
GI System Presentation
Most patients with nonclassic CF will maintain adequate pancreatic function. However in classic CF patients, steatorrhea, or greasy stools, is typically present that can lead to a failure to thrive, resulting in malnutrition. Infants and small children will show an increase in frequency of small stools. Newborns may present with meconium ileus, which is considered diagnostic of CF. Older patients may experience constipation, abdominal cramping, and flatulence. This presentation is due to the obstruction of the pancreatic ducts and intestinal tract and their inability to digest essential nutrients.
Pancreatic malfunction can also lead to an insulin deficiency, which is often a later finding detected by a loss in weight, an increase in blood glucose levels, and a failed oral glucose tolerance test (OGTT).
Reproductive Presentation
As patients reach adolescent and adult ages, tests may show azoospermia due to blockage of or the congenital bilateral absence of the vas deferens. Females may experience reduced fertility as cervical fluids have lower water content and decreased thinning during ovulation.9,15,16
DIAGNOSIS
As of 2010, all states were required to perform CF newborn screening.17 The screening test checks for immunoreactive trypsinogen (IRT), a chemical produced by the pancreas. The IRT tends to be high in babies with CF. A second test may be done with a follow-up IRT test or a DNA test to look for a genetic mutation that causes CF. The CF newborn screening consists of a test called the quantitative pilocarpine iontophoresis sweat test (QPIT). The QPIT came about due to the risk of hyperpyrexia associated with older methods that utilized plastic body bags to make patients sweat. QPIT uses only a small area on the forearm, which is then stimulated to secrete sweat through the skin by iontophoresis of pilocarpine. Sweat from the stimulated area is then collected and analyzed for chloride content. Chloride concentrations are quantified as: normal: ≤39 mmol/L; intermediate: 40 to 59 mmol/L; and abnormal: ≥60 mmol/L. Values ≥60 mmol/L are consistently diagnostic of CF. It is suggested that samples from two sites will increase the reliability of the diagnosis3,7 (Fig. 18-4).
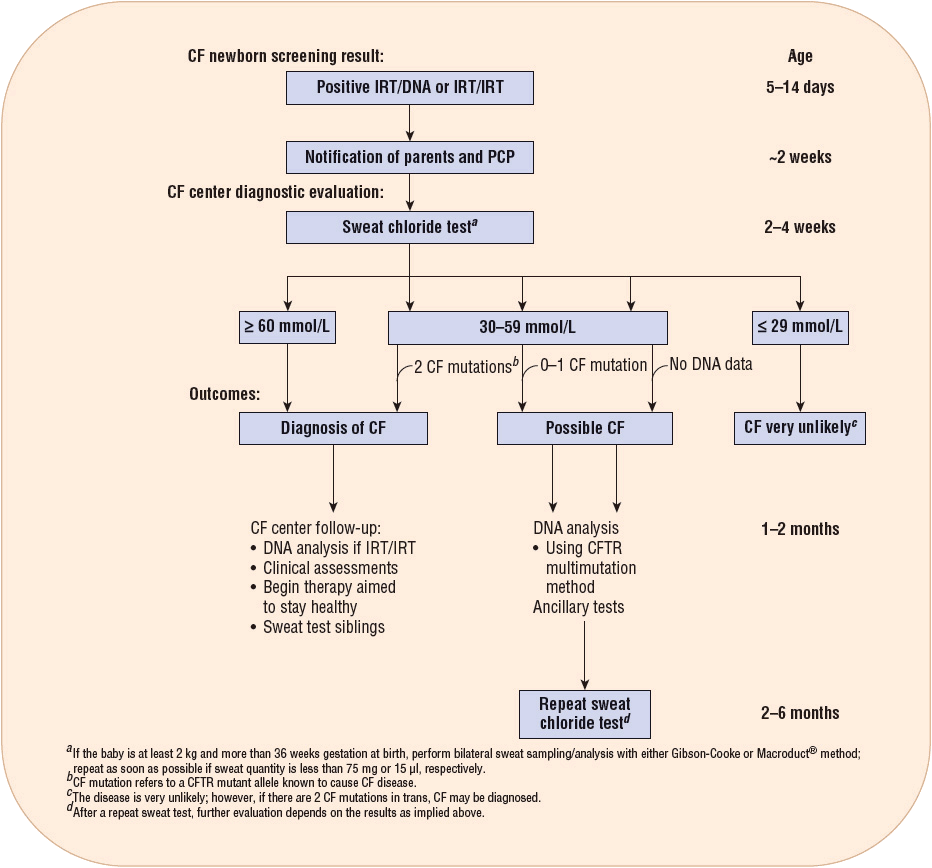
FIGURE 18-4 The CF diagnostic process for screened newborns. (From Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for the diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation Consensus Report. J Pediatr 2008;153(2):S4–S14.)
DESIRED OUTCOMES
Pharmacists play a vital role in assisting patients to reach the following long- and short-term goals. Since CF affects multiple organ systems, there are several therapeutic goals that must be addressed for each system.8
Sinopulmonary
1. Prevent and treat sinusitis.
2. Increase FEV1 and promote optimal pulmonary function tests and prevent pulmonary exacerbations.
a. Promote effective airway clearance by providing counseling on the use of appropriate medications and chest physiotherapy.
b. Prevent and treat colonization of the lungs with pathogens.
c. Prevent and treat acute exacerbations.
GI
1. Control pancreatic insufficiency by providing adequate enzyme supplementation.
2. Optimize growth and nutritional status.
3. Promote healthy bowel habits.
4. Maintain normal fat-soluble vitamin levels.
Reproduction
1. Provide mutation analysis with appropriate genetic counseling at the time of diagnosis and periodically thereafter.
Psychosocial
1. Keep these patients living essentially normal lives by being active in school and the workplace.
2. Encourage compliance with pharmacological and nonpharmacological therapies in order to help prolong CF patient’s lives.
NUTRITION
![]() In healthy individuals, the pancreas is vital to the absorption and digestion of essential nutrients for the body’s growth and function. In pancreatic-insufficient CF individuals, the resulting inability to absorb these nutrients may lead to malnourishment. The focus of treatment lies in achieving and maintaining normal weight for adults and normal growth patterns for children. This is mostly achieved by managing GI and pulmonary symptoms, monitoring nutrient and energy intakes, and addressing psychosocial and financial issues. Numerous population-based studies have provided strong evidence to support optimization of nutritional status, due to its association with an improved pulmonary function. The CF Foundation recommends that both children and adults maintain normal nutritional status, due to its association with healthy pulmonary function, including a better FEV1, and an increase in survival.
In healthy individuals, the pancreas is vital to the absorption and digestion of essential nutrients for the body’s growth and function. In pancreatic-insufficient CF individuals, the resulting inability to absorb these nutrients may lead to malnourishment. The focus of treatment lies in achieving and maintaining normal weight for adults and normal growth patterns for children. This is mostly achieved by managing GI and pulmonary symptoms, monitoring nutrient and energy intakes, and addressing psychosocial and financial issues. Numerous population-based studies have provided strong evidence to support optimization of nutritional status, due to its association with an improved pulmonary function. The CF Foundation recommends that both children and adults maintain normal nutritional status, due to its association with healthy pulmonary function, including a better FEV1, and an increase in survival.
To help meet this desired outcome, the CF Foundation recommends energy intakes greater than the standard for the general population to support weight gain and maintenance in children over 2 years and in adults. Trial evidence gathered from population-based studies has shown that energy intakes of 110% to 200% compared to the general health population intakes yield improved nutritional status in CF individuals. The CF Foundation has also established consensus-based assessment parameters to monitor nutritional status in CF individuals. These parameters and goals are listed in Table 18-2. In order to achieve these goals, pancreatic enzyme replacement therapy (PERT) is utilized to improve fat absorption due to pancreatic insufficiency. For patients who consistently fail to meet weight requirements, the clinician must consider the use of nutritional supplements that may be given orally or enterally via a percutaneous endoscopic gastrostomy (PEG) tube.
TABLE 18-2 Cystic Fibrosis Foundation Nutritional Assessment Parameters and Recommendations

PERT has been proven both safe and efficacious in improving nutritional status in CF patients and is recommended in addition to adequate dietary intake. Consensus-based guidelines have established a dose of 500 to 2500 lipase units per kilogram (kg) of body weight per meal; or 10,000 units per kg per day; or 4,000 units per gram of dietary fat per day. Generic enzyme supplements are not bioequivalent; therefore, the CF Foundation does not recommend their use.
Until recently, pancreatic enzymes were considered nutritional supplements, and were not under the FDA jurisdiction. New regulations now require all pancreatic enzyme supplements to obtain FDA approval. Table 18-3 shows currently used enzyme preparations.18–39
TABLE 18-3 Pancreatic Enzyme Supplements
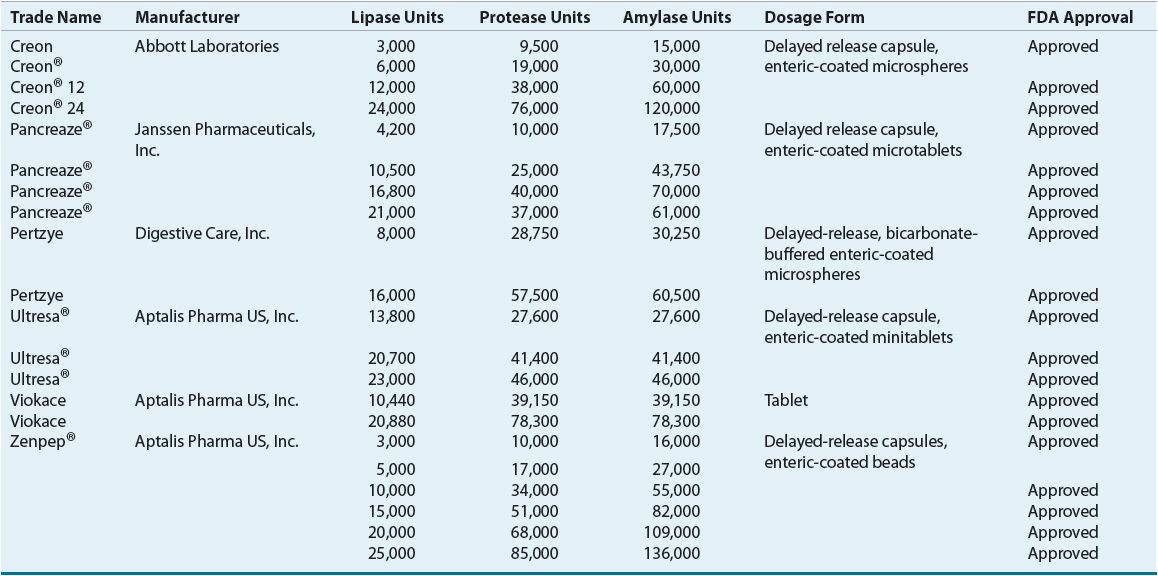
Most preparations are capsules containing enteric-coated microspheres or enteric-coated tablets designed to withstand the acidic environment in the stomach allowing for absorption in the small intestine. Frequently CF patients require the addition of histamine receptor antagonists or proton-pump inhibitors in order to create an alkaline environment in the intestine. Enteric-coated capsules should not be crushed but may be opened and mixed with nonalkaline food. However, if allowed to sit in food for a prolonged amount of time, the enteric coating will be lost and enzymes inactivated. Enzymes are administered prior to meals and snacks.40
Patients dosed beyond the recommended guidelines may develop fibrosing colonopathy, which leads to colonic strictures. This condition should be considered in individuals who have evidence of obstruction, bloody diarrhea, or ascites, as well as in patients who have a combination of abdominal pain, ongoing diarrhea, and/or poor weight gain. Risk factors for fibrosing colonopathy include: age <12 years; enzyme dosages >6,000 lipase units/kg/meal for more than 6 months; history of meconium ileus or distal intestinal obstruction syndrome (DIOS); history of any intestinal surgery; and inflammatory bowel disease.
Patients who experience fibrosing colonopathy are treated by reducing the dose of enzyme supplements, or with oral laxatives and/or enemas, all of which have been proven effective. More severe cases may require surgical intervention.41
BONE HEALTH AND VITAMIN SUPPLEMENTATION
Increased longevity in CF patients has revealed bone disease as an emerging complication. Many studies have observed that 50% to 75% of CF adults have low bone density and increased rates of fractures. CF patients are especially at risk as a result of several contributing factors: malabsorption of vitamin D, poor nutritional status, physical inactivity, glucocorticoid therapy, delayed pubertal maturation, and early hypogonadism. Increased bone resorption and decreased bone formation are likely stimulated by elevated serum cytokine levels triggered by chronic pulmonary inflammation. Additionally, chronic infections lead to bone loss in patients regardless of pancreatic sufficiency. Pancreatic insufficient CF patients have the inability to absorb fat-soluble vitamins A, D, E, and K (ADEK). Decreased calcium absorption and intake can also compound this problem. As bone disease progresses, this can lead to exclusion from lung transplantation, which is often a life-saving operation for individuals with CF.
Appropriate bone density monitoring for CF patients requires obtaining levels of fat-soluble vitamins yearly, as well as treatment with daily supplementation. Special multivitamin formulations contain high amounts of fat-soluble vitamins designed to deliver the appropriate doses required. Recommended vitamin D levels are a minimum of 30 ng/mL (75 nmol/L). Even with these precautions, adequate vitamin D levels may be difficult to maintain due to altered absorption, reduced fat mass, and minimal exposure to sun light. Medical management of CF patients can also contribute to bone disease by the administration of glucocorticoids, posttransplant immunosuppressant therapies, and antibiotic therapies that require protection from sunlight exposure.42–51
PULMONARY HEALTH AND TREATMENT
![]() One of the fundamentals of pulmonary care in CF patients is airway clearance. CF patients, in general, have impaired mucociliary clearance that results in thick sputum, predisposing them to chronic infections and inflammation. Effective airway clearance involves the use of a bronchodilator, a mucolytic medication, and chest percussion. It is recommended that airway clearance therapy (ACT) be initiated within the first few months of life. Table 18-4 shows typical medications used in airway clearance.
One of the fundamentals of pulmonary care in CF patients is airway clearance. CF patients, in general, have impaired mucociliary clearance that results in thick sputum, predisposing them to chronic infections and inflammation. Effective airway clearance involves the use of a bronchodilator, a mucolytic medication, and chest percussion. It is recommended that airway clearance therapy (ACT) be initiated within the first few months of life. Table 18-4 shows typical medications used in airway clearance.
TABLE 18-4 Airway Clearance Therapies
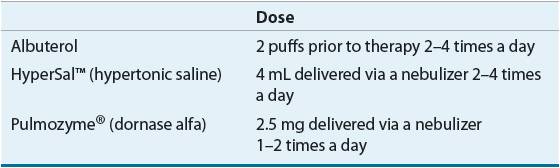
Choosing a particular ACT routine for a patient is based on the patient’s needs. There is no consensus on the optimal method of ACT. The regimen including duration or number of treatments per day may be changed in response to acute illness or exacerbations.
Chest percussion was originally performed by hand, with a cupped hand pounding on the chest that generates percussion or vibration. Currently, the most convenient method is the use of a percussion vest. Aerobic exercise is also effective and recommended for improved airway clearance.52
The recommended sequence of clearance therapy or “pulmonary toilet” regimen is as follows (note that these therapies are recommended for individuals ≥6 years of age and are administered concurrently with percussion therapy):
1. Bronchodilator: Albuterol is commonly used for this indication. It helps open up the airways and prevents bronchospasm.
2. Hypertonic saline (HyperSal®): It hydrates the airway mucus secretions and facilitates mucociliary function.
3. Dornase alfa (Pulmozyme®): Enzyme that cleaves extracellular DNA, which results in decreased viscosity of mucus.
4. Aerosolized antibiotics (i.e., Aztreonam [Cayston], tobramycin [TOBI®]): If this therapy is indicated based on severity of lung disease and sputum cultures, it is administered after the CF patient completes percussion therapy.
Bronchodilator therapy is recommended for patients ≥6 years of age who demonstrate bronchiole hyperresponsiveness or a bronchodilator response. Chronic use of bronchodilator therapy is recommended to improve lung function by enhancing mucociliary action.53,54
Inhaled hypertonic saline is a novel agent used for the treatment of CF. Based on the “low-volume model” theory, the use of hypertonic saline would restore airway hydration and enhance mucociliary function.55 Hypertonic saline is recommended for patients ≥6 years of age. A study conducted in Australia showed that CF patients who surfed had better pulmonary outcomes than other patients who did not surf.56 Researchers believed that the inhalation of ocean water helped to improve FEV1 in CF patients that surfed. In this study, 24 patients were randomly assigned to receive a daily treatment of 7% hypertonic saline with or without pretreatment of a control. Clearance and pulmonary function were measured during a 14-day period. Results showed significant improvement in FEV1 and FVC, as well as improvement of respiratory symptoms in hypertonic saline patients. The study also demonstrated that these patients were able to sustain mucus clearance for >8 hours. Other studies assessing the use of hypertonic saline have supported this study, showing an improvement in lung function and a 56% reduction in exacerbations.53–57
Dornase alfa (Pulmozyme®) is also recommended in all patients ≥6 years of age, and is strongly recommended in patients with moderate-to-severe lung disease, to improve lung function and reduce exacerbations. Three randomized controlled trials and a crossover trial involving 520 patients were conducted. Study results showed improvement in FEV1 by 3.2% and a reduction in exacerbations.53,58,59
Antiinflammatory Therapies
Pulmonary inflammation begins early in life, as shown by the predominance of proinflammatory mediators that can be seen on bronchiolar lavage. A normal inflammatory response to bacteria becomes pathologic in CF patients who have both a prolonged and exaggerated reaction. Treatment of this inflammatory response is crucial to treating the CF patient.
Antiinflammatory therapies must address the neutrophil response and inhaled therapies will target the endobronchial location, which is the site of inflammation. Using medications that terminate the inflammatory process may be effective. Airway clearance and antibiotics will help control the inflammatory stimulation. Steroids and nonsteroidal antiinflammatory drugs (NSAIDs) are not widely used due to long-term safety concerns. High-dose ibuprofen (20 to 30 mg/kg of body weight twice daily) has proven efficacious in a study where patients showed less decline in pulmonary function when compared to patients given placebo. Patients on high dose ibuprofen were able to maintain weight and had less hospital admissions. The benefits of this regimen exceed the risks of GI complications and nephrotoxicity. Despite these outcomes, less than 10% of CF patients in the United States are on this regimen. The low number of patients utilizing this proven therapy may be related to the requirement to obtain a specific therapeutic level of ibuprofen, which in turn requires frequent blood draws for pharmacokinetic monitoring.53,60–63
Studies with macrolides have shown an inhibition of the neutrophil migration and a decrease in production of proinflammatory mediators. It is unclear at this point if the antiinflammatory effects of macrolides are a combination of antimicrobial and/or immunomodulatory mechanisms of action. A study conducted in Japan first demonstrated the benefit of macrolides against Pseudomonas aeruginosa. Four randomized controlled trials have since demonstrated this effect with azithromycin (250 to 500 mg) given three times weekly, which has led to increased nutritional status and decreased pulmonary infections. Other treatments are under investigation, but larger studies are needed before they become recommended therapies.53,60,64,65
Infectious Disease
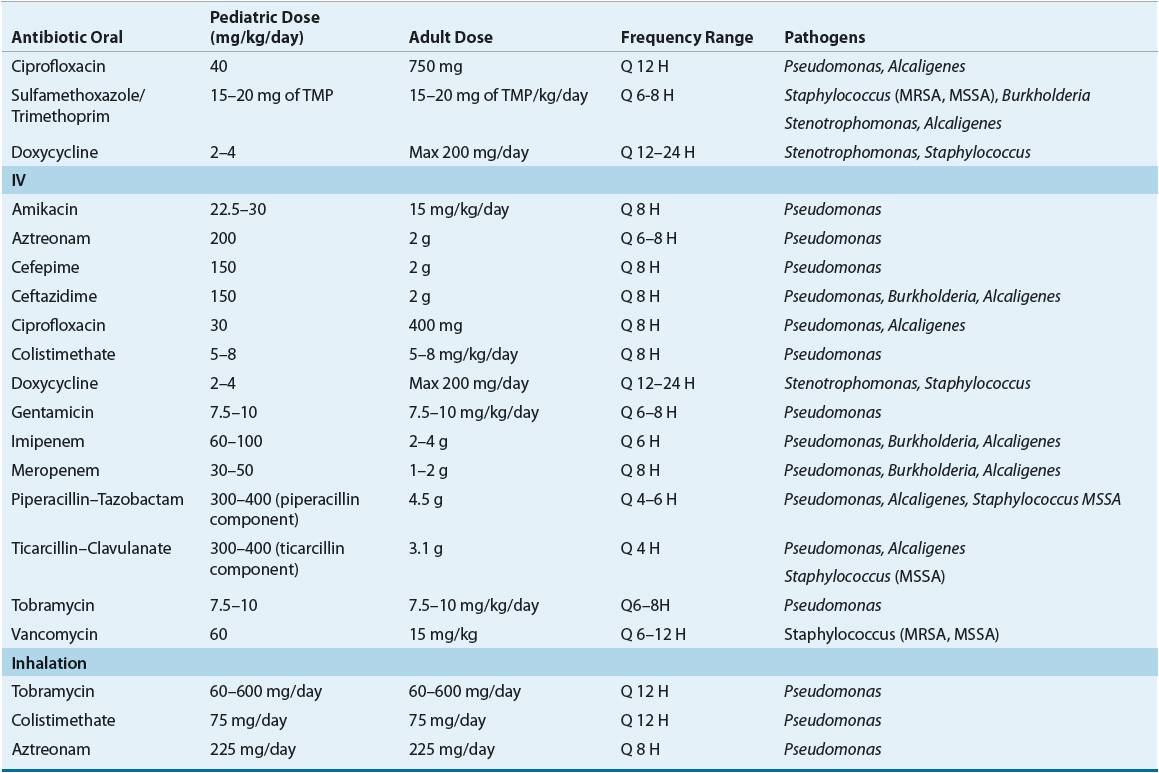
![]() Antibiotic therapy plays two integral roles in the treatment of CF patients: improving pulmonary function and preventing pulmonary failure. Oral, IV, and aerosolized antibiotic formulations are indicated and utilized in patients who experience acute pulmonary exacerbations, are chronically infected with P. aeruginosa, or require prevention of chronic P. aeruginosa infection. A major disadvantage of treatment in CF patients is that pathogens are not fully eradicated from the airways and will often develop resistance. Unfortunately, this limits antimicrobial selection, and can contribute to deterioration of pulmonary function (Table 18-5).
Antibiotic therapy plays two integral roles in the treatment of CF patients: improving pulmonary function and preventing pulmonary failure. Oral, IV, and aerosolized antibiotic formulations are indicated and utilized in patients who experience acute pulmonary exacerbations, are chronically infected with P. aeruginosa, or require prevention of chronic P. aeruginosa infection. A major disadvantage of treatment in CF patients is that pathogens are not fully eradicated from the airways and will often develop resistance. Unfortunately, this limits antimicrobial selection, and can contribute to deterioration of pulmonary function (Table 18-5).
TABLE 18-5 Antimicrobial Agents Utilized in CF