Drug-Induced Kidney Disease
KEY CONCEPTS
![]() The initial diagnosis of drug-induced kidney disease (DIKD) typically involves detection of elevated serum creatinine and blood urea nitrogen, for which there is a temporal relationship between the toxicity and use of a potentially nephrotoxic drug.
The initial diagnosis of drug-induced kidney disease (DIKD) typically involves detection of elevated serum creatinine and blood urea nitrogen, for which there is a temporal relationship between the toxicity and use of a potentially nephrotoxic drug.
![]() DIKD is best prevented by avoiding the use of potentially nephrotoxic agents for patients at increased risk for toxicity. However, when exposure to these drugs cannot be avoided, recognition of risk factors and specific techniques, such as hydration, may be used to reduce potential nephrotoxicity.
DIKD is best prevented by avoiding the use of potentially nephrotoxic agents for patients at increased risk for toxicity. However, when exposure to these drugs cannot be avoided, recognition of risk factors and specific techniques, such as hydration, may be used to reduce potential nephrotoxicity.
![]() Acute tubular necrosis is the most common presentation of DIKD in hospitalized patients. The primary agents implicated are aminoglycosides, radiocontrast media, cisplatin, amphotericin B, and osmotically active agents.
Acute tubular necrosis is the most common presentation of DIKD in hospitalized patients. The primary agents implicated are aminoglycosides, radiocontrast media, cisplatin, amphotericin B, and osmotically active agents.
![]() Angiotensin-converting enzyme inhibitors and nonsteroidal antiinflammatory drugs are associated with hemodynamically mediated kidney injury, the pathogenesis of which is a decrease in glomerular capillary hydrostatic pressure.
Angiotensin-converting enzyme inhibitors and nonsteroidal antiinflammatory drugs are associated with hemodynamically mediated kidney injury, the pathogenesis of which is a decrease in glomerular capillary hydrostatic pressure.
![]() Acute allergic interstitial nephritis is observed in up to 27% of kidney biopsies performed for hospitalized patients with unexplained acute kidney injury. Clinical manifestations of AIN typically present approximately 14 days after initiation of therapy and include fever, maculopapular rash, eosinophilia, arthralgia, often with pyuria, hematuria, proteinuria, and oliguria.
Acute allergic interstitial nephritis is observed in up to 27% of kidney biopsies performed for hospitalized patients with unexplained acute kidney injury. Clinical manifestations of AIN typically present approximately 14 days after initiation of therapy and include fever, maculopapular rash, eosinophilia, arthralgia, often with pyuria, hematuria, proteinuria, and oliguria.
INTRODUCTION
Numerous diagnostic and therapeutic agents have been associated with the development of drug-induced kidney disease (DIKD) or nephrotoxicity. It is a relatively common complication with variable presentations depending on the drug and clinical setting, inpatient or outpatient. Manifestations of DIKD include acid–base abnormalities, electrolyte imbalances, urine sediment abnormalities, proteinuria, pyuria, and/or hematuria. However, the most common manifestation of DIKD is a decline in the glomerular filtration rate (GFR), which results in a rise in serum creatinine (Scr) and blood urea nitrogen (BUN) and several other indicators of acute and chronic kidney injury (see eChap. 18 and Chap. 28).1 Initial diagnosis of DIKD is often delayed as it typically is based on the detection of elevated Scr and BUN, for which there is a temporal relationship between the kidney injury and exposure to the potentially nephrotoxic drug. This is consistent with classic qualitative definitions of acute renal failure, which have relied on either an abrupt increase in Scr or an abrupt decline in urine output (see eChap. 18 and Chap. 28). Historically, the clinical use of numerous definitions of acute renal failure and nephrotoxicity based on quantitative changes in the Scr concentration and other clinical end points made it extraordinarily difficult to ascertain their true incidence.2 During the last decade however, standard terminology (e.g., acute kidney injury, AKI) and diagnostic criteria based on a combination of physiologic measurements (e.g., Scr and urine output) have been adopted and are now routinely used in clinical practice and research.3,4 This will likely lead to new epidemiologic data about DIKD that are more accurate.
Nephrotoxicity is often reversible if one discontinues the use of the offending agent, but in some cases there may still be an AKI and progression to stage 5 chronic kidney disease (CKD), which includes end-stage renal disease (ESRD). Currently, many different mechanisms are responsible for the pathogenesis of DIKD, and the introduction of new drugs with novel mechanisms of action provides the potential for the identification of new presentations of AKI and CKD. This chapter reviews the epidemiology, pathophysiology, risk factors, and basic principles of prevention of DIKD. Detailed discussions of these issues plus management strategies are presented for the most commonly used agents that have been associated with a moderate to high likelihood of DIKD.
EPIDEMIOLOGY
The incidence and characteristics of outpatient or community-acquired DIKD are not well understood since mild toxicity is often unrecognized in this setting. However, the acquisition of data regarding the pharmacoepidemiology of these effects has become more important as care increasingly shifts to the outpatient setting. The incidence of community-based AKI that required dialysis was recently reported to be 29.5 per 100,000 person years and 522.4 per 100,000 person years for patients not requiring dialysis.5 Although the incidence of drug-induced AKI was not specifically reported, earlier studies have implicated community-acquired DIKD in up to 20% of hospital admissions due to AKI.6 Conversely, AKI has been reported in up to 7% of hospitalized patients,7 and as many as 20% to 30% of critically ill patients may experience AKI during their hospitalization.8,9 Drug-induced causes have been implicated in up to 60% of all cases of in-hospital AKI and as such are a recognized source of significant morbidity and mortality. Although the incidence of in-hospital antibiotic-induced AKI alone has been reported to be as high as 36%, it appears to be declining, while cases of in-hospital AKI due to nonselective nonsteroidal antiinflammatory drugs (NSAIDs), angiotensin-converting enzyme inhibitors (ACEIs), chemotherapeutic agents, and antiviral drugs are increasing.1
CLINICAL PRESENTATION Drug-Induced Kidney Disease
![]() Because the most common manifestation of DIKD is a decline in GFR leading to a rise in Scr and BUN, the onset of toxicity in hospitalized, acutely ill patients is most often recognized by routine laboratory monitoring. Decreased urine output may also be an early sign of toxicity, particularly with radiographic contrast media, NSAIDs, and ACEIs. In the outpatient setting, nephrotoxicity is often recognized by the development of symptoms such as malaise, anorexia, vomiting, volume overload (shortness of breath or edema), and hypertension. Scr or BUN concentrations and urine collection for creatinine clearance may subsequently be measured to quantify the degree of decline in GFR. Marked intrasubject between-day variability of Scr values have been noted (±20% for values within the normal range; see eChap. 18). Furthermore, they may be altered as the result of dietary changes and initiation of drug therapy, which may interfere with the assay procedure. Thus changes in Scr or urine output consistent with the diagnostic criteria for AKI (see Chap. 28),10 when correlated temporally with the initiation of drug therapy, are a common threshold for the identification of DIKD.
Because the most common manifestation of DIKD is a decline in GFR leading to a rise in Scr and BUN, the onset of toxicity in hospitalized, acutely ill patients is most often recognized by routine laboratory monitoring. Decreased urine output may also be an early sign of toxicity, particularly with radiographic contrast media, NSAIDs, and ACEIs. In the outpatient setting, nephrotoxicity is often recognized by the development of symptoms such as malaise, anorexia, vomiting, volume overload (shortness of breath or edema), and hypertension. Scr or BUN concentrations and urine collection for creatinine clearance may subsequently be measured to quantify the degree of decline in GFR. Marked intrasubject between-day variability of Scr values have been noted (±20% for values within the normal range; see eChap. 18). Furthermore, they may be altered as the result of dietary changes and initiation of drug therapy, which may interfere with the assay procedure. Thus changes in Scr or urine output consistent with the diagnostic criteria for AKI (see Chap. 28),10 when correlated temporally with the initiation of drug therapy, are a common threshold for the identification of DIKD.
Nephrotoxicity may also be evidenced by primary alterations in renal tubular function without a corresponding loss of glomerular filtration. In this setting, urinary enzymes and low-molecular-weight proteins may be used as earlier and more specific biomarkers of nephrotoxicity compared with Scr and BUN, which are relatively insensitive markers of kidney injury.11,12 Scr and BUN are used as surrogates of kidney function, not injury per se, and typically significant kidney injury must have occurred days before a rise in either is evident. Urinary excretion of kidney injury molecule-1 (KIM-1), N-acetyl-β-glucosaminidase, γ-glutamyl transpeptidase, glutathione S-transferase, neutrophil gelatinase-associated lipocalin (NGAL), and interleukin-18 are markers of proximal tubular injury and have been used for the early detection of acute kidney damage in several patient populations.11–14 For example, the transmembrane protein KIM-1 is upregulated for patients with ischemic acute tubular necrosis (ATN), appearing in the urine within 12 hours after the ischemic insult.14 Urinary N-acetylglucosamine (NAG) concentrations are a highly sensitive indicator of AKI and have been shown to detect AKI in critically ill patients up to 4 days prior to a rise in Scr was observed.14 Similarly, urinary NGAL is an early marker of AKI, preceding a rise in Scr by up to 3 days. In the future, urinary biomarkers such as KIM-1, NAG, and NGAL may facilitate the earlier detection of kidney injury and diagnosis of nephrotoxicity and minimize the long-term consequences of this common drug-induced disorder.11,12,14
PRINCIPLES FOR PREVENTION OF DRUG-INDUCED NEPHROPATHY
![]() The primary principle for prevention of DIKD is to avoid the use of nephrotoxic agents for patients at increased risk for toxicity. Therefore, an awareness of potentially nephrotoxic drugs and knowledge of risk factors that increase renal vulnerability is essential.15 Exposure to these drugs often cannot be avoided, so several interventions have been proposed to reduce the potential for the development of nephrotoxicity, for example, adjustment of medication dosage regimens based on accurate estimates of kidney function, and careful and adequate hydration to establish high urine flow rates.16 Other preventative strategies are still theoretical and/or investigational and relate directly to the specific nephrotoxic mechanisms of a given drug.
The primary principle for prevention of DIKD is to avoid the use of nephrotoxic agents for patients at increased risk for toxicity. Therefore, an awareness of potentially nephrotoxic drugs and knowledge of risk factors that increase renal vulnerability is essential.15 Exposure to these drugs often cannot be avoided, so several interventions have been proposed to reduce the potential for the development of nephrotoxicity, for example, adjustment of medication dosage regimens based on accurate estimates of kidney function, and careful and adequate hydration to establish high urine flow rates.16 Other preventative strategies are still theoretical and/or investigational and relate directly to the specific nephrotoxic mechanisms of a given drug.
The several specific drug-induced renal structural–functional alterations that are responsible for the vast majority of cases of DIKD are listed in Table 31-1. This chapter discusses the pathophysiologic mechanisms responsible for the development of DIKD with these agents in detail, along with clinical presentation, prevention strategies, therapeutic management approaches, and relevant monitoring plans.
TABLE 31-1 Drug-Induced Kidney Structural–Functional Alterations

TUBULAR EPITHELIAL CELL DAMAGE
![]() Drugs that lead to renal tubular epithelial cell damage typically do so via direct cellular toxicity or ischemia. Damage is most often localized in the proximal and distal tubular epithelia and is termed acute tubular necrosis when cellular degeneration and sloughing from proximal and distal tubular basement membranes are observed.17 This classically manifests as cellular debris-filled, muddy-brown, granular casts in the urinary sediment.17,18 Specific indicators of proximal tubular injury include metabolic acidosis with bicarbonaturia; glycosuria in the absence of hyperglycemia; and reductions in serum phosphate, uric acid, potassium, and magnesium as a result of increased urinary losses. Indicators of distal tubular injury include polyuria from failure to maximally concentrate urine (i.e., nephrogenic diabetes insipidus), metabolic acidosis from impaired urinary acidification, and hyperkalemia from impaired potassium excretion.
Drugs that lead to renal tubular epithelial cell damage typically do so via direct cellular toxicity or ischemia. Damage is most often localized in the proximal and distal tubular epithelia and is termed acute tubular necrosis when cellular degeneration and sloughing from proximal and distal tubular basement membranes are observed.17 This classically manifests as cellular debris-filled, muddy-brown, granular casts in the urinary sediment.17,18 Specific indicators of proximal tubular injury include metabolic acidosis with bicarbonaturia; glycosuria in the absence of hyperglycemia; and reductions in serum phosphate, uric acid, potassium, and magnesium as a result of increased urinary losses. Indicators of distal tubular injury include polyuria from failure to maximally concentrate urine (i.e., nephrogenic diabetes insipidus), metabolic acidosis from impaired urinary acidification, and hyperkalemia from impaired potassium excretion.
Acute Tubular Necrosis
ATN is the most common presentation of DIKD in the inpatient setting. The primary agents associated with this type of injury are aminoglycosides, radiocontrast media, cisplatin, amphotericin B, foscarnet, and osmotically active agents such as immunoglobulins, dextrans, and mannitol.9,19
Aminoglycoside Nephrotoxicity
Incidence Aminoglycoside antibiotic-associated nephrotoxicity has been reported to occur in between 10% and 25% of patients receiving a therapeutic course.9,20,21 Critically ill patients appear to have a higher risk for nephrotoxicity with reported rates as high as 58%.20 The large variance is in part a result of the use of different definitions of toxicity, variability between agents in the class, and the risk factors present in the study population.
Clinical Presentation Clinical evidence of aminoglycoside-associated nephrotoxicity is typically seen within 5 to 10 days after initiation of therapy and manifests as a gradual progressive rise in Scr and BUN and decrease in creatinine clearance.9 Patients usually present with nonoliguria, that is, they maintain urine volumes greater than 500 mL/day and sometimes have microscopic hematuria and proteinuria.9,19 Although renal magnesium wasting can occur (i.e., daily excretion of more than 10 to 30 mg), the risk of symptomatic hypomagnesemia is generally low. Full recovery of kidney function is common if aminoglycoside therapy is discontinued immediately upon discovering signs of toxicity.11 However, severe AKI may develop occasionally, and for these individuals renal replacement therapy may be required (see Chap. 28). The diagnosis of aminoglycoside-associated nephrotoxicity is often difficult, particularly in critically ill patients with multiple comorbidities and is confounded by other factors that are independently associated with the development of AKI.20 For instance, concurrent dehydration, sepsis, hypotension, ischemia, and use of other nephrotoxic drugs frequently contribute to AKI in patients who are receiving aminoglycosides.17
Pathogenesis Aminoglycoside-associated ATN is primarily due to accumulation of high drug concentrations within proximal tubular epithelial cells, and subsequent generation of reactive oxygen species that produce mitochondrial injury, which leads to cellular apoptosis and necrosis.10,19 This results in cell sloughing from proximal tubular basement membranes into the tubular lumen, which can result in tubular obstruction and back leakage of the glomerular filtrate across the damaged tubular epithelium. Toxicity is related to cationic charge of the drugs in this class, which facilitates their binding to negatively charged renal tubular epithelial membrane phospholipids in the proximal tubules, followed by intracellular transport and concentration in lysosomes. The number of cationic groups on the drug molecule appears to correlate with the degree of nephrotoxicity, which is consistent with the observation of higher rates of toxicity with neomycin versus gentamicin, followed by tobramycin, then amikacin.9,15
Risk Factors Multiple risk factors for aminoglycoside-associated nephrotoxicity have been identified: the aggressiveness of aminoglycoside dosing, synergistic toxicity as the result of combination drug therapy, and preexisting clinical conditions of the patient (Table 31-2).9,15,20
TABLE 31-2 Potential Risk Factors for Aminoglycoside Nephrotoxicity

Prevention Aminoglycoside-associated ATN may be prevented by careful and cautious selection of patients and the use of alternative antibiotics whenever possible and as soon as microbial sensitivities are known. Commonly used alternatives include fluoroquinolones (e.g., ciprofloxacin or levofloxacin) and third-or fourth-generation cephalosporins (e.g., ceftazidime or cefepime). When aminoglycosides are necessary, gentamicin, tobramycin, and amikacin are most commonly used, but therapy should be selected to optimize antimicrobial efficacy.22 Furthermore, it is imperative to avoid volume depletion, limit the total aminoglycoside dose administered, and avoid concomitant therapy with other nephrotoxic drugs. Future therapeutic alternatives may include new aminoglycoside congeners that retain the desired bactericidal activity and yet are devoid of nephrotoxicity, and may also include concurrent use of antioxidant compounds such as vitamin E and N-acetylcysteine.23,24
Prospective, individualized pharmacokinetic monitoring has been used for more than three decades, and its use has been associated with a decrease in the incidence of aminoglycoside-associated nephrotoxicity.25 These studies, however, were often small and statistically underpowered. High-dose intermittent dosing of aminoglycosides, termed once daily dosing, used in combination with other antibiotics, has been intensively investigated as a practical cost-effective method to maintain antimicrobial efficacy while reducing the risk of AKI.24,26,27 The reduction in incidence may be the result of limited proximal tubular aminoglycoside uptake during the transient, high-peak serum concentrations, and because of the presence of low aminoglycoside concentrations for a greater proportion of the dosing interval, which facilitates excretion of the aminoglycoside.22 Although greater clinical efficacy and reduced nephrotoxicity may be realized with once daily compared with standard dosing, seriously ill, immunocompromised, and elderly patients, as well as those with preexisting kidney disease, are not ideal candidates for this approach.24
Management Aminoglycoside use should be discontinued or the dosage regimen revised if AKI is evident (i.e., there is an Scr increase of 0.5 mg/dL [44 μmol/L] or more that is not attributable to another cause). Other nephrotoxic drugs should be discontinued if possible, and the patient should be maintained adequately hydrated and hemodynamically stable.26 Short-term renal replacement therapy may be necessary, but ESRD has rarely been reported to be solely the result of aminoglycoside toxicity.28
Radiographic Contrast Media Nephrotoxicity
Incidence The incidence of radiographic contrast media-induced nephrotoxicity (CIN) has declined over the past decade from approximately 15% to 7% of all patients receiving iodinated contrast; yet it remains the third leading cause of hospital-acquired AKI, accounting for up to 11% of cases.29 The incidence varies depending on the population studied and presence of risk factors; rising from <2% for patients with normal kidney function up to 50% for patients with CKD or diabetes mellitus.30 As the number of risk factors associated with CIN increases, there is a proportional increase in the incidence of nephrotoxicity and in hospital and postdischarge mortality rates.29,31 A 5.5-fold increased risk of death has been reported for patients who develop CIN compared with those who do not, with the highest mortality rates observed for patients who developed AKI and required renal replacement therapy.19,21 In-hospital and 2-year mortality rates of 36% and 81%, respectively, have been reported for patients who developed CIN and those that required dialysis. An in-hospital mortality rate of only 7% was observed in those with CIN not requiring dialysis.29
Clinical Presentation CIN is usually transient in nature, presenting most commonly as nonoliguria with kidney injury apparent within the first 24 to 48 hours after the administration of contrast. The Scr concentration usually peaks between 3 and 4 days after exposure, with recovery after 7 to 10 days.32,33 However, irreversible oliguric (urine volume <500 mL/day) AKI requiring dialysis has been reported in high-risk patients.31 Urinalysis typically reveals tubular enzymuria with hyaline and granular casts but may also be completely void of casts. The urine sodium concentration and fractional excretion of sodium are frequently low, with the latter typically <1% (<0.01).
Pathogenesis The primary mechanisms by which contrast media induces nephrotoxicity are renal ischemia and direct cellular toxicity.29 Renal ischemia likely results from systemic hypotension and simultaneous acute vasoconstriction caused by disruption of normal prostaglandin synthesis and the release of adenosine, endothelin, and other renal vasoconstrictors. Subsequently, a 50% sustained reduction in renal blood flow that lasts for several hours immediately following contrast administration may be evident.29 This reduced renal blood flow leads to increased concentrations of contrast in the renal tubules and exacerbates the direct cytotoxicity. The extent of cellular toxicity is directly related to the duration of tubular cell exposure to contrast. Thus, preservation of high urinary flow rates with adequate hydration before, during, and after contrast administration is vital to keep renal blood flow as high as reasonably possible to minimize tubular cell exposure to the contrast agent.29 In humans, plasma osmolality is normally between 275 and 290 mOsm/kg (275 and 290 mmol/kg). Since low- and high-osmolar contrast agents are hyperosmolar to plasma (i.e., 600 to 800 mOsm/kg [600 to 800 mmol/kg] and ~2,000 mOsm/kg [~2,000 mmol/kg], respectively), their use may result in osmotic diuresis, dehydration, renal ischemia, and increased blood viscosity caused by red blood cell aggregation. Oxidative stress has also been implicated in the development of ATN after contrast administration,34 which may explain the possible benefit of the antioxidant N-acetylcysteine.35
Risk Factors Decreased renal blood flow exacerbates the ischemic and direct cytotoxic effects of contrast media on the renal tubules. Therefore, preexisting kidney disease, particularly in those with estimated GFR <60 mL/min/1.73 m2, is the most important risk factor present in up to 60% of patients who develop CIN.29 Other patient-specific risk factors include conditions associated with decreased renal blood flow (i.e., congestive heart failure, dehydration/volume depletion, and hypotension), and patients with atherosclerosis and reduced effective circulating arterial blood volume appear to also have an elevated risk.33 Diabetes is also a significant risk factor, likely due to coexisting kidney disease (diabetic nephropathy). The presence of multiple myeloma has traditionally been considered a relative contraindication for contrast use, but the risk appears to be associated with concomitant dehydration, kidney disease, or hypercalcemia rather than the diagnosis itself. Larger volumes or doses of contrast and the use of low- as well as high-osmolar contrast agents are also independent predictors of CIN. Intraarterial administration of contrast confers greater risk than IV administration.29 Lastly, concurrent use of nephrotoxins and drugs that alter renal hemodynamics such as NSAIDs and ACEIs also increases risk. Risk factors are additive, and there is a proportional increase in the incidence of CIN and associated mortality as the number of risk factors increases.29,31
Prevention CIN can be anticipated in the majority of patients who are at risk; so the use of preventative procedures is justified for virtually all patients. Table 31-3 lists the recommended interventions for prevention of contrast nephrotoxicity. All patients scheduled to receive contrast media should be assessed for risk factors, and the risk-to-benefit ratio should be considered.29,33,36,37 High-risk patients can be identified by evaluating medical history and indication for the contrast study, along with their most recent Scr concentrations. Nephrotoxicity is best prevented in high-risk patients by using alternative imaging procedures (e.g., ultrasound, noncontrast magnetic resonance imaging, and nuclear medicine scans).36 However, if contrast media must be used, the smallest adequate volume should be administered. If the ratio of the volume of contrast to be infused relative to the patient’s creatinine clearance is ≥3.7 (≥222 if creatinine clearance is expressed in units of milliliters per second), the likelihood of nephrotoxicity is markedly increased.38 Therefore, in general, the volume of contrast administered should not be greater than twice the baseline estimated creatinine clearance.29
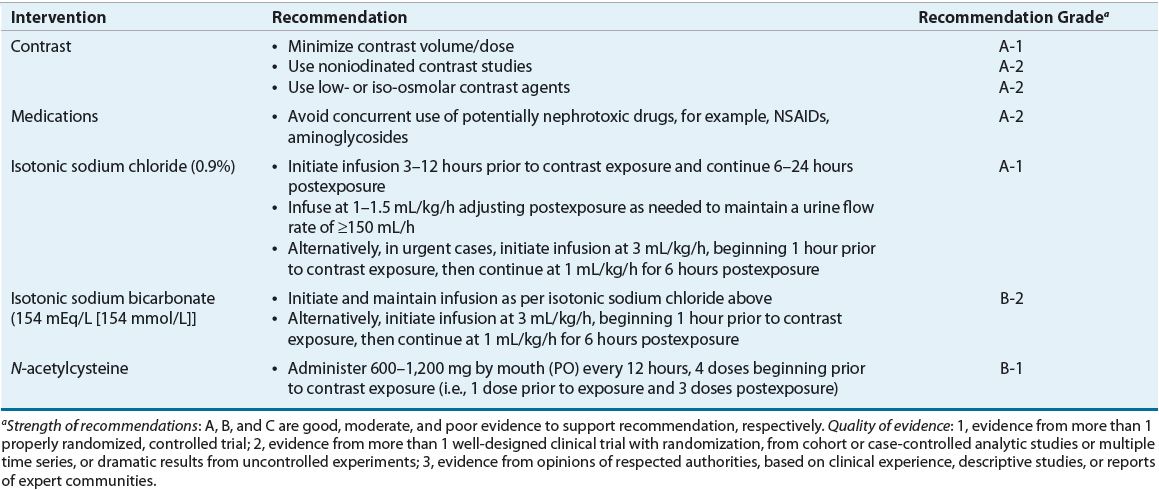
Low-osmolar (600 to 800 mOsm/kg; 600 to 800 mmol/kg) nonionic (iohexol and iopamidol) and ionic (ioxaglate) contrast agents may be used to minimize the incidence of nephrotoxicity. Standard hyperosmolar contrast media (e.g., low- and high-osmolar agent) are not reabsorbed in the kidney and cause osmotic diuresis, which contributes to the renal toxicity observed with these agents. Low-osmolar contrast agents have less than half the osmolality of high-osmolar (~2,000 mOsm/kg; ~2,000 mmol/kg) agents and are associated with less toxicity, especially when used for patients with preexisting kidney disease.39 However, use of low-osmolar agents does not preclude the development of nephrotoxicity. Even low-osmolar agents are hyperosmolar relative to plasma, which is likely the reason they are associated with greater nephrotoxicity than the iso-osmolar nonionic contrast agent iodixanol. Iodixanol has been shown to have the lowest risk for CIN for patients with CKD and diabetes.29,39
Clinical Controversy…
Volume expansion and correction of dehydration prior to contrast administration is a mainstay of preventive therapy.36,40 Parenteral hydration with isotonic saline before and after contrast administration reduces the incidence of toxicity, particularly in high-risk patients, and is currently the most widely accepted preventative intervention.40 Volume expansion may exert its beneficial effects through dilution of contrast media, prevention of renal vasoconstriction leading to ischemia, preservation of high urine flow rates, decreased tubular cell exposure to contrast, and avoidance of tubular obstruction. Hydration with isotonic sodium bicarbonate has been shown to provide more protection than saline, perhaps by reducing the formation of pH-dependent oxygen free radicals,41 but recent studies reported contradictory findings.36,42 Larger, adequately powered studies are needed to confirm these findings and to demonstrate conclusively that bicarbonate-based hydration is superior to saline. The use of oral hydration regimens has also been proposed but requires further study to clarify its role and is not currently recommended in lieu of parenteral hydration.40
N-acetylcysteine is a thiol-containing antioxidant that may effectively reduce the risk of developing CIN for patients with preexisting kidney disease. Despite the publication of dozens of clinical trials and meta-analyses, a therapeutic benefit of NAC has not been consistently demonstrated, and its therapeutic role remains controversial.43 Nevertheless, its use should be considered, along with hydration, for all patients who are at high risk of toxicity.35,37,43 The recommended N-acetylcysteine dosing regimen for prevention of CIN is to give four doses of 600 mg to 1,200 mg orally every 12 hours, with the first dose administered prior to contrast exposure (see Table 31-3).29,35,41 Finally, other nephrotoxic drugs should be discontinued if possible, and subsequent contrast studies appropriately timed to minimize cumulative toxicity.
Clinical Controversy…
Renal replacement therapy, including intermittent hemodialysis and continuous modalities, for example, continuous venovenous hemofiltration (CVVH), effectively removes iodinated contrast, and was considered by some to be a therapeutic option for the prevention of CIN.44 However, because of the logistical issues (e.g., technical difficulty), high cost of renal replacement therapy, and lack of consistent clinical efficacy data, currently this approach is not recommended.44,45
Management Currently there is no specific therapy available for managing established CIN. Care is supportive as described in Chapter 28. Kidney function (e.g., Scr and urine output), electrolytes (e.g., sodium and potassium), and volume status should be closely monitored.
Cisplatin Nephrotoxicity
Incidence Cisplatin is one of the most important and widely used antineoplastic drugs for the treatment of solid tumors, often demonstrating exceptional efficacy (i.e., cure rates over 90% in testicular cancers).46,47 Unfortunately, the primary dose-limiting toxicity of platin-containing compounds is nephrotoxicity. Cisplatin nephrotoxicity occurs in 20% to 30% of patients and is a significant cause of morbidity.46,48 Carboplatin, a second-generation platinum analog, is associated with a lower incidence of nephrotoxicity than cisplatin and thus is the preferred agent in high-risk patients.49
Clinical Presentation Cisplatin administration results in impaired tubular reabsorption and decreased urinary concentration ability, leading to increased excretion of salt and water (i.e., polyuria) within 24 hours of treatment. Polyuria persists, and a decrease in GFR evidenced by a rise in Scr concentration may be seen within 72 to 96 hours after cisplatin administration.47,48 Scr peaks approximately 10 to 14 days after initiation of therapy, with recovery by 21 days. As many as 25% of patients may have reversible elevations in Scr and BUN for 2 weeks after cisplatin treatment. However, kidney damage is dose related and cumulative with subsequent cycles of therapy, so the Scr concentration may continue to rise, and irreversible kidney injury may result.48,49 Hypomagnesemia is a hallmark finding of cisplatin nephrotoxicity, due to impaired magnesium reabsorption and thus increased urinary losses.47 Hypomagnesemia is often accompanied by hypocalcemia and hypokalemia and may be severe, leading to seizures, neuromuscular irritability, or personality changes. Urinalysis typically reveals leukocytes, renal tubular epithelial cells, and granular casts.48
Pathogenesis The pathogenesis of cisplatin nephrotoxicity is multifactorial in nature and likely begins with cellular uptake and accumulation of the drug in proximal tubular epithelial cells to concentrations that may reach five times the serum concentration.46,48 Tubular cell exposure to cisplatin then activates a series of cell signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway, p53, caspase, and the generation of reactive oxygen species, that collectively promote tubular cell injury and death via necrosis and/or apoptosis.49 Simultaneous production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) within tubular cells activates an inflammatory response, which may worsen the renal insult.46 Although tubular damage is evident in both the proximal and distal segments, the majority occurs in the proximal tubules and is followed by a progressive loss of glomerular filtration capacity and impaired distal tubular function. Renal biopsies generally reveal necrosis of proximal and distal tubules and collecting ducts, with no obvious morphological changes to the glomeruli.48
Risk Factors Risk factors include increased age, dehydration, renal irradiation, concurrent use of nephrotoxic drugs, large cumulative doses, and alcohol abuse.47,48
Prevention The best renoprotective strategy is a combination of interventions, including prospective dose reduction and decreased frequency of administration, which usually requires using the platin compounds in combination with other chemotherapeutic agents, avoiding concurrent use of other nephrotoxic drugs, and ensuring patients are euvolemic or somewhat hypervolemic prior to initiating treatment.46,50 Vigorous hydration with isotonic saline should be used for all patients with a goal of maintaining at least 100 to 150 mL/h of urine output during and after cisplatin treatment. Hydration should be initiated 12 to 24 hours prior to and continued for 2 to 3 days after cisplatin administration at rates of 100 to 250 mL/h, as tolerated, to maintain a urine flow of 3 to 4 L/day.47
Amifostine, an organic thiophosphate that is converted to an active metabolite, chelates cisplatin in normal cells and reduces the nephrotoxicity, neurotoxicity, ototoxicity, and myelosuppression associated with cisplatin and carboplatin therapy. It is also thought to serve as a thiol donor, thereby reducing intracellular reactive oxygen species and corresponding oxidative stress that plays a critical role in the development of cellular injury.23,46 Pretreatment with amifostine should be considered for patients who are at high risk for kidney injury, particularly patients who are elderly, volume depleted, have CKD, or are receiving other nephrotoxic drugs concurrently. The current recommended dose of amifostine is 910 mg/m2 administered IV over 15 minutes, beginning 30 minutes prior to cisplatin administration.51 Common toxicities include acute hypotension, nausea, and fatigue.
Other renoprotective strategies include the use of hypertonic saline (e.g., administration of each dose in 250 mL of 3% saline) to reduce tubular cisplatin uptake. Classic antioxidants such as ascorbic acid, thiol-based antioxidants such as α-lipoic acid and N-acetylcysteine, which reduce oxidative damage by acting as a sulfhydryl donor, and the disulfiram metabolite diethyldithiocarbamate to reduce cytochrome P450 2E1-mediated generation of hydroxyl radicals have also been evaluated.52 Finally, reduced renal exposure can be achieved with the use of localized intraperitoneal administration in conjunction with systemic administration of sodium thiosulfate for those with peritoneal tumors.47
Management AKI caused by cisplatin therapy is usually partially reversible with time and supportive care, including dialysis. Kidney function indices should be closely followed, with Scr and BUN concentrations checked daily. Serum magnesium, potassium, and calcium concentrations should be monitored daily and corrected as needed.48,49 Hypocalcemia and hypokalemia may be difficult to reverse until hypomagnesemia is corrected. Progressive kidney disease caused by cumulative nephrotoxicity may be irreversible and in some cases may lead to ESRD and require chronic dialysis support.
Amphotericin B Nephrotoxicity
Incidence Variable rates of amphotericin B nephrotoxicity have been reported that correspond in large part to the cumulative dose administered. Nephrotoxicity may be seen in nearly 30% of patients receiving median cumulative doses as low as 240 mg and reaches an incidence of >80% when cumulative doses approach 5 g.53,54 Although numerous studies demonstrate lower rates of nephrotoxicity with liposomal formulations compared with conventional amphotericin B, it is difficult to compare rates of toxicity between products and studies because of the variability in the study populations, doses administered, and inconsistent definitions of nephrotoxicity and methods of assessment.55,56
Clinical Presentation Dose-dependent nephrotoxicity is often evident after administration of cumulative doses of 2 to 3 g as non-oliguria, renal tubular potassium, sodium, and magnesium wasting, impaired urinary concentrating ability, and distal renal tubular acidosis.9 Although the cumulative dose is a significant risk factor, the time to onset of kidney injury varies considerably, ranging from a few days to weeks.53 Tubular dysfunction usually manifests 1 to 2 weeks after treatment is begun, and potassium and magnesium replacement may be necessary. This is typically followed by a decrease in GFR and a rise in Scr and BUN concentrations. Consequently, kidney function indices should be closely followed, with Scr and BUN concentrations checked daily, and serum magnesium, potassium, and calcium concentrations monitored every other day and corrected as needed.
Pathogenesis Amphotericin B nephrotoxicity occurs predominantly via two mechanisms. The first is direct tubular epithelial cell toxicity resulting from interaction of amphotericin B with ergosterol in the cell membrane, leading to increased tubular cell membrane permeability, lipid peroxidation, and eventual necrosis of proximal tubular cells.53 Tubular injury appears to be exacerbated by ischemic injury, which is a result of a reduction in renal blood flow and GFR due to afferent arteriolar vasoconstriction.9,53
Risk Factors Risk factors that impact the likelihood of developing amphotericin B nephrotoxicity include preexisting kidney disease, large individual and cumulative doses, short infusion times, volume depletion, hypokalemia, increased age, and concomitant administration of diuretics and other nephrotoxins (cyclosporine in particular).9,53
Prevention Permanent decrements in GFR are best prevented by incorporating a low threshold (i.e., if Scr reaches 2 mg/dL [177 μmol/L] on 2 consecutive days) for stopping amphotericin B or switching to a liposomal formulation. Several lipid formulations of amphotericin B (e.g., amphotericin B lipid complex, liposomal amphotericin B) are available and should be used in most high-risk patients as they reduce nephrotoxicity by enhancing drug delivery to sites of infection and reducing interaction with tubular epithelial cell membranes.53,56 Nephrotoxicity can also be minimized by limiting the cumulative dose, increasing the infusion time, ensuring the patient is well hydrated, and avoiding concomitant administration of other nephrotoxins.9 Administration of 1 L IV 0.9% sodium chloride daily during the course of therapy appears to reduce toxicity and a single infusion of saline 10 to 15 mL/kg prior to administration of each dose of amphotericin B are generally recommended.53 A number of other antifungal agents such as itraconazole, voriconazole, and caspofungin are viable alternatives and are now routinely used in lieu of amphotericin B for patients at high risk of developing nephrotoxicity.9



