1Abbreviations: Cys, cysteine (any form), with thiol and disulfide forms indicated as CySH, CySSCy, and CySSR; cyst(e)ine, Cys or cystine; EAR, estimated average requirement; Glu, glutamate; Gly, glycine; GSH, glutathione; Hcy, homocysteine (any form), with thiol and disulfide forms indicated as HcySH, HcySSHcy, and HcySSR; homocyst(e)ine, Hcy or homocystine; H2S, hydrogen sulfide; Km, Michaelis-Menten constant; Met, methionine; NHANES III, Third National Health and Nutrition Examination Survey; RDA, recommended dietary allowance; SAA, sulfur amino acid; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; TauT, taurine transporter; tCys, sum of all forms of plasma Cys including those present as thiol, half-disulfide, mixed disulfide, and protein-bound disulfide; tHcy, sum of all forms of Hcy; THF, tetrahydrofolate; TPN, total parenteral nutrition; UL, tolerable upper intake level.
Cysteine (Cys) is a sulfur-containing amino acid, whereas taurine is a product of Cys oxidation, and homocysteine (Hcy) is a metabolite of methionine (Met), which also serves as a precursor of Cys sulfur. The importance of sulfur amino acids (SAAs) for growth or protein synthesis was first recognized in 1915, when Osborne and Mendel (1) demonstrated that the addition of cystine to a low-casein diet resulted in restoration of rapid growth of rats. Womack et al. (2) demonstrated that cyst(e)ine was not essential for rats when dietary Met was adequate and that the effect of cyst(e)ine resulted from its ability to replace part, but not all, of the Met in the diet. Rose and Wixom (3) demonstrated the same relationship between Met and cyst(e)ine requirements in their studies of amino acid requirements of men. Thus, only Met is considered an essential amino acid, but in practice the Met or total SAA requirement is usually met by a combination of Met and cyst(e)ine. N-Acetylcysteine, which is readily deacetylated to Cys, is used clinically in the treatment of acetaminophen overdose and for prevention of radiocontrast-induced nephropathy.
During the last decades of the twentieth century, the nutritional importance of taurine and the clinical significance of Hcy were recognized. Taurine is an end product of Cys catabolism. It was first isolated from the bile of the ox (Box taurus) in 1827 (4). Interest in taurine surged following the discovery in 1975 that cats fed diets containing little or no taurine suffered retinal degeneration accompanied by low retinal and plasma taurine concentrations (5). This finding was soon followed by the observation that infants fed purified formulas lacking taurine had lower plasma and urine taurine levels than did infants fed pooled human milk (6, 7). As a consequence of increasing evidence of a possible role of taurine in development, taurine has been added to most human infant formulas since the mid-1980s. Numerous possible therapeutic applications of taurine have been suggested, including treatment of patients with hypertension, cardiovascular disease, diabetes, hepatic disorders, chronic renal failure, sepsis, and inflammatory disorders.
Hcy, a metabolite of Met and precursor of the sulfur atom in Cys biosynthesis, was discovered by du Vigneaud in 1932 (8) as the product of Met demethylation. The role of homocyst(e)ine in the conversion of Met sulfur to Cys (the transsulfuration pathway by which the sulfhydryl group of Hcy replaces the hydroxyl group of serine to form Cys) was studied in the years to follow, and investigators showed that homocyst(e)ine could support the growth of animals fed diets deficient in Cys, Met, or choline. Homocystinuria, an inborn error of metabolism, was identified in 1962 when individuals with mental retardation were screened for abnormal urinary amino acid patterns (9). Subsequently, investigators recognized that small increases in plasma Hcy concentrations were associated with folate, vitamin B12, or vitamin B6 deficiency and with increased risk of cardiovascular disease, neural tube defects, and various other diseases found in the general population (10, 11, 12, 13).
CHEMISTRY, NOMENCLATURE, AND CELLULAR/EXTRACELLULAR FORMS
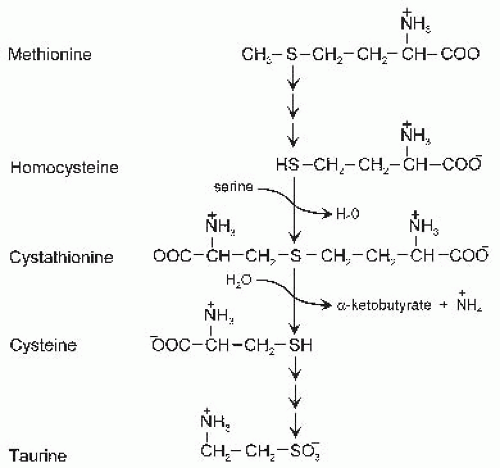
The structures of Cys, Hcy, and taurine and their relations with precursor amino acids (Met and serine) are shown in Figure 33.1. As in other amino acids with an asymmetric carbon atom, the L-isomers of Met, Hcy, and Cys are the biologically active forms. Both Hcy and Cys have a free sulfhydryl group. The carbon skeleton of Hcy, which is derived from Met, has one more carbon than does the carbon chain of Cys, which is derived from serine. Taurine, 2-aminoethane sulfonate, is formed from Cys by removal of the carboxyl group and oxidation of the sulfur to form a sulfonic acid group. The carboxyl (pKa ˜1.7), sulfonic (pKa ˜1.5), sulfhydryl (pKa ˜8.3), and amino (pKa ˜9 to 11) groups all undergo ionization; the zwitterionic forms shown in Figure 33.1 are the dominant species at physiologic pH.
DIETARY CONSIDERATIONS, TYPICAL INTAKES, AND RECOMMENDED INTAKES
The body’s need for Cys must be met by the diet and may be supplied either as preformed cyst(e)ine or as its sulfur precursor Met. The carbon skeleton for cyst(e)ine biosynthesis is provided by serine, which can be synthesized in the body. Under most circumstances, sufficient taurine can apparently be synthesized from the SAAs, but taurine may be classified as conditionally essential under some conditions. Little Hcy is present in the diet, but it is formed in the process of Met metabolism in the body. No dietary requirement exists for Hcy intake.
Fig. 33.1. Structures and metabolic relations of sulfur amino acids.
Methionine and Cyst(e)ine
The SAAs, Met and Cys, are normally consumed as components of dietary proteins. Normal Western diets provide approximately 2 to 4 g/day of SAAs for adults (14). Based on the Third National Health and Nutrition Examination Survey (NHANES III; 1988 to 1994), the mean Met intake of 31- to 50-year-old men and women was 2.3 ± 0.04 and 1.6 ± 0.2 (standard error [SE]) g/day, or 15.4 and 10.7 mmol/day, respectively. Mean Cys intake for the same age group was 1.3 ± 0.02 and 0.89 ± 0.01 g/day, or 10.7 and 7.4 mmol/day, respectively. Thus, mean total SAA intake was 26.1 and 18.1 mmol/day, respectively, for men and women. SAAs tend to be more abundant in animal and cereal proteins than in legume proteins, and the ratio of Met to Cys tends to be higher in animal proteins than in plant proteins (Table 33.1).
For adults, the current estimated average requirement (EAR) for Met plus Cys intake is 15 mg · kg-1 · day, and the recommended dietary allowance (RDA) is 19 mg · kg-1 · day (14). Considering that approximately one third of the SAA requirement on a weight basis is taken in as Cys rather than Met, the current RDA is consistent with the estimated safe intake of Met (21 mg · kg-1 · day-1) reported by Di Buono et al (15) but less than the estimated safe intakes (25 mg · kg-1 · day-1) determined by Young et al (16) and Storch et al (17). The current EAR for protein intake is 0.66 g · kg-1 · day, and the RDA is 0.8 g · kg-1 · day-1. Thus, a desirable amino acid pattern for adults includes at least 24 mg Met plus Cys per gram of protein (i.e., 19 mg/0.8 g = 24 mg/g). Mixtures of proteins consumed in the United States actually contain a higher proportion of SAAs, approximately 35 mg Met plus Cys per gram of protein. The RDA for SAAs (1.3 g/day for a 70-kg adult) is easily met in diets commonly consumed in the United States. Even the lowest Met plus Cys intakes reported in NHANES III (1st percentile; 1.87 for men and 1.4 g for women in the 31- to 50-year age bracket) exceeded the current RDA (14).
The current RDA for SAAs for pregnant and lactating women is 25 and 26 mg · kg-1 · day-1. For infants and children, the SAA RDA is 43 mg · kg-1 · day-1 for 7- to 12-month-old infants, 28 mg · kg-1 · day-1 for 1- to 3-year-old children, 22 mg · kg-1 · day-1 for 4- to 8-year-old children, 22 and 21 mg · kg-1 · day-1 for 9- to 13-year-old boys and girls, and 21 and 19 mg · kg-1 · day-1 for 14- to 18-year-old boys and girls (14). The Institute of Medicine did not establish a tolerable upper intake level (UL) for Cys or Met intake because data were insufficient for dose-response assessment and derivation of a UL for healthy adults.
TABLE 33.1 METHIONINE AND CYSTEINE CONTENT OF SELECTED FOODS
AMOUNT
PATTERN
METHIONINE
CYSTEINE
METHIONINE
CYSTEINE
FOOD
(mg/100 g Edible Portion)
(mg/g Protein)
Cheese, cheddar
652
125
26
5
Milk, whole
83
30
25
9
Egg, whole, chicken
392
289
32
24
Chicken, flesh only, cooked roasted
800
370
28
13
Beef, round, separable lean only
557
224
26
11
Wheat flour, whole meal
186
278
14
21
Corn grits, regular, dry
196
237
22
22
Oats, regular, dry
266
398
17
25
Peanut butter
292
365
10
13
Soybean, green, cooked
150
113
12
9
Brown rice, dry
142
152
19
21
Despite the availability of food proteins that provide ample amounts of SAAs, it is likely that some individuals have inadequate intakes, either because of inadequate intake of total protein or because of selection of a restricted variety of proteins that provide inadequate SAAs. Analysis of diets of long-term vegans living in California indicated an average protein intake of 64 g/day and an SAA intake of 1.04 g (7.6 mmol)/day (18); this is equivalent to an intake of approximately 15 mg · kg-1 · day-1 of SAAs and an amino acid pattern of 16 mg Met plus cyst(e)ine per gram of protein. This level of intake would meet the EAR, but not the RDA, for SAAs. Adults with higher-than-average requirements would be at risk of inadequate intake. Careful selection of plant proteins to ensure an adequate intake of SAAs may be important for individuals consuming strict vegan diets.
Mixtures of proteins typically consumed in the United States supply approximately 40% of the total SAAs as Cys and 60% as Met on a molar basis. This distribution would seem to allow optimal use of SAAs based on estimates of approximately 50% for the ability of Cys to replace Met in the diet. In cases of limited ability to convert Met to Cys (whether from hepatic dysfunction, inborn errors of Met metabolism to Cys, or prematurity), the total amount of SAAs in the diet, the balance of Cys and Met, and the adequacy of taurine should be considered.
Taurine
Taurine is not considered to be an essential nutrient because it is an end product of SAA metabolism. Nevertheless, a considerable amount of taurine may be obtained from the diet. Food taurine content has not been widely determined, but data from several reports (19, 20, 21, 22) are summarized in Table 33.2. Taurine is present in most animal foods and is either absent or present in very low levels in most plant foods. Relatively high concentrations of taurine have been reported for some lower plants such as seaweeds (22).
Consistent with the large range of taurine content of foods, the taurine content of typical diets varies widely. Analysis of the diets of strict vegans living in England yielded no detectable taurine, whereas the diets of omnivores contained 463 ± 156 (SE) µmol/day (23). The analyzed taurine intake of adults fed omnivorous diets in a clinical study center in the United States was 1000 to 1200 µmol/day (19). In a cross-sectional study involving 24 populations in 16 countries (24), the highest median urinary taurine levels were found in adults in Beppu, Japan (2181 and 1590 µmol/day for men and women, respectively), whereas the lowest median urinary excretions of taurine were observed in men in St. John, Canada (192 µmol/day) and women in Moscow, Russia (128 µmol/day). The large variation in taurine excretion largely reflects the range of dietary intake of animal foods, especially of seafood.
TABLE 33.2 TAURINE CONTENT OF SELECTED FOODS
FOOD
TAURINE CONTENT
Animal foods
Poultry
89-2,245 µmol/100 g wet weight
Beef and Pork
307-489 µmol/100 g wet weight
Processed meats
251-981 µmol/100 g wet weight
Seafood
84-6,614 µmol/100 g wet weight
Cow’s milk
18-20 µmol/100 mL
Yogurt, ice cream
15-62 µmol/100 mL
Cheese
Not detected
Plant foods
Most fruits, vegetables, seeds, cereals, grains, beans, peanuts
Not detected
Soybeans, chickpeas, black beans, pumpkin seeds, some nutsa
≤1-4 µmol/100 g wet weight
Seaweeds (marine algae)
1.5-100 µmol/100 g wet weight
a Low reported values should be regarded as upper limits because contamination of food or methodologic interference by compounds that coelute with taurine could account for these low concentrations of taurine.
Values from Laidlaw et al. (19), Pasantes-Morales et al. (20), Roe and Weston (21), and Kataoka and Ohnishi (22).
Taurine-supplemented beverages have been popular for decades in Japan, where Taisho Pharmaceutical’s Lipovitan is a favored drink. Since the 1990s, these taurine-supplemented beverages have become increasingly popular in many other countries, including the United States. Examples of taurine-supplemented energy drinks include Red Bull, Dark Dog, Monster, and Rockstar, which contain 1000 mg (8000 µmol) per 240- or 250-mL can. Clearly, consumption of taurine-supplemented beverages dramatically increases taurine intake to eight or more times the typical intakes in a population. Nevertheless, little reason exists to conclude that the amounts of taurine in these beverages have either therapeutic benefits or adverse effects.
Breast-fed infants receive taurine from their mother’s milk. The taurine content of milk from lactating women was reported to be 413 ± 71 (SE) µmol/L for early milk (1 to 7 days) and 337 ± 28 µmol/L for later milk (>7 days) (25, 26). The mean taurine concentration of milk from lactating lacto-ovo vegetarian women is only slightly lower than that of omnivores (26). The mean taurine content of milk of lactating vegan women is lower than that of lactating omnivores, but values between the two groups overlap considerably, and the taurine concentration in milk of vegan mothers is approximately 30 times the level in the cow’s milk-based infant formulas used before the mid-1980s (23).
Because strict vegan diets tend to be lower in total SAA content and virtually free of taurine, individuals consuming vegan diets are at somewhat greater risk of inadequate SAA status. Adult humans who consume a strict vegetarian diet have been reported to have lower plasma taurine concentrations and greatly reduced urinary taurine excretion compared with omnivores. Vegans consuming little or no preformed taurine are healthy, however, and the children born to and nursed by vegan mothers appear to have normal growth and development (23).
Nevertheless, by general consensus, taurine is considered to be conditionally essential during infant development and probably for adults in some special circumstances. Because the brain and retina of human infants are not fully developed at birth and may be vulnerable to the effects of taurine deprivation, it has been judged prudent to supplement human infant formulas and pediatric feeding solutions with taurine (7, 27). During the 1980s, manufacturers of infant formulas began adding taurine to their products, and currently taurine is added to virtually all human infant formulas and pediatric parenteral solutions throughout the world (28). Taurine is added to infant formulas at levels comparable to those in human milk or at somewhat higher levels in formulas for premature infants (19).
ABSORPTION, TRANSPORT, AND EXCRETION
Intestinal Absorption
Absorption of the products of protein digestion across the intestinal epithelium is highly efficient (˜95% to 99%). Dietary Met, a precursor of Cys, is transported by neutral amino acid transport systems B0,+ (SLC6A14) and L (SLC7A8 + SLC3A2), and as Met-containing peptides by the peptide transport system (PEPT1), across the luminal (brush-border) membrane of enterocytes. Met can exit enterocytes into the interstitial fluid through the alanine, serine, and Cys (asc) preferring system (SLC7A10 + SLC3A2). Dietary Cys is absorbed as CySH, CySSCy, and as Cys-containing peptides by a variety of L-amino acid and peptide transport systems in the small intestinal mucosa. Transport of Cys (CySH) is accomplished by the sodium (Na+)-dependent neutral amino acid transporter system B (SLC6A19) in the apical membrane and by the Na+-independent system asc (SLC7A10 + SLC3A2) in the basolateral membrane of the intestinal mucosa cells. Cystine (CySSCy) is transported by system B0,+ (SLC7A9 + SLC3A1) and x–c (SLC7A11 + SLC3A2), both Na+-independent systems that are present in the apical membranes of the intestinal mucosa (29, 30).
Efficient absorption of taurine is facilitated by two apical membrane transporters: the β-amino acid or taurine transporter (TauT; SLC6A6), which is a Na+ and chloride (Cl–)-dependent carrier that serves taurine, β-alanine, and γ-aminobutyric acid; and the hydrogen ion (H+)-coupled transporter PAT1 (SLC36A1) that may be important only when taurine intakes are very high (31). Efflux of taurine from the enterocyte across the basolateral membrane may also be mediated by TauT (32). The intestinal uptake of taurine and the expression of the TauT in the intestine do not respond to the level of dietary SAAs or taurine (33).
The reabsorption of the taurine-conjugated bile acids secreted into the lumen of the bile occurs in the ileum, and this enterohepatic reabsorption plays an important role in taurine conservation. Apical uptake of luminal bile acids is mediated by the Na+-dependent bile acid transporter ASBT (SLC10A2) in the distal ileum, whereas efflux across the basolateral membrane may result from the heterodimeric organic solute transporter Ostα-Ostβ (34).
Blood Transport and Intracellular Forms
The small intestinal cells use dietary SAAs for protein and glutathione (GSH) synthesis and also have the capacity for SAA catabolism (35). Amino acids enter the plasma and circulate as free amino acids until they are removed by tissues. The disulfide forms of Cys (protein-bound Cys, PSSCy, and cystine, CySSCy) dominate in the more oxidized extracellular environment. The plasma membranes of cells in tissues have various amino acids transporters, similar to those in the small intestine, that take up Cys from the plasma. System xc– is up-regulated in response to oxidative stress or amino acid deficiency and allows uptake of more cystine to facilitate GSH and protein synthesis (36, 37, 38).
The liver removes a substantial proportion of the SAAs from the portal circulation and uses them for synthesis of protein and GSH or for catabolism to taurine and sulfate. GSH is exported into plasma, and this Cys-containing tripeptide as well as its metabolites, CysGly and γ-GluCys, can be a source of Cys to tissues. Most of the Cys in cells exists in peptide (GSH) or polypeptide/protein forms. For free Cys, the thiol form of Cys (CySH) dominates intracellularly.
Plasma Hcy levels are normally low because Hcy is not normally present in the diet and only very small amounts are normally released from tissues into the plasma. Intracellularly, low concentrations of Hcy are present in free (HcySH) and protein-bound (PSSHcy) forms. Extracellularly, Hcy is present predominantly as mixed disulfides of Hcy with protein (PSSHcy) or Cys (HcySSCy).
Physiologic and Genetic Factors Influencing Use or Production of Cysteine, Homocysteine, and Taurine
Cyst(e)ine Transport
Cys uptake can be diminished and its loss from the plasma increased by defects in cystine transport. Cystinuria is an inherited disorder of cystine and dibasic amino acid transport by the system b0,+ transporter that is expressed by the kidney and small intestine (39, 40, 41). Because other intestinal amino acid and peptide transporters are not affected, these amino acids are generally absorbed from the intestine in sufficient amounts. The defect in the renal transporter results in elevated levels of lysine, ornithine, arginine, and cystine in the urine, however, because of the lack of reabsorption of these amino acids by the proximal tubular cells of the kidney (42). The major complication of cystinuria is the formation of cystine kidney stones because cystine is a highly insoluble amino acid with an aqueous solubility limit of 250 mg/L (1 mmol/L).
In another genetic disorder of cystine transport, cystinosis, cystine reutilization is prevented, and this leads to the accumulation of cystine in lysosomes. In cystinosis, mutations in the gene for cystinosin give rise to the lack of a functional lysosomal cystine transporter (43). This situation causes cystine from degraded proteins to accumulate inside the lysosomes of cells and leads to tissue damage. Malfunctioning kidneys and corneal crystals are the main initial features of the disorder. Patients with cystinosis are usually treated by administration of the thiol cysteamine to reduce intracellular cystine. Cysteamine enters the lysosome and reacts with cystine to form Cys and a Cyscysteamine disulfide, which are both able to leave the lysosome through other transport systems.
Methionine Metabolism to Homocysteine and Cysteine
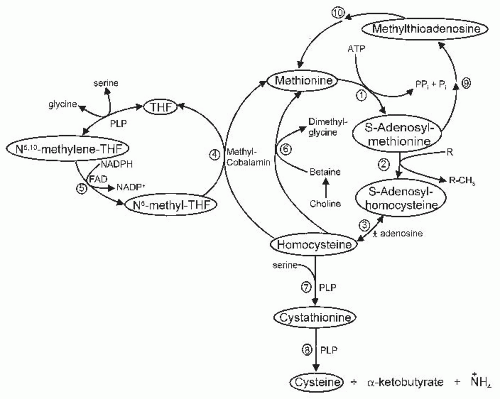
Because Cys can be synthesized in the body from Met (Hcy) sulfur and serine, Cys levels can be affected by Met intake and by various factors that influence Met metabolism, including the pathways for Met transmethylation, Hcy remethylation, and Hcy transsulfuration, which are summarized in Figure 33.2. Met is metabolized by formation of S-adenosylmethionine (SAM), transfer of the methyl group to various substrates forming S-adenosylhomocysteine (SAH), and the hydrolysis of SAH to form Hcy. Thus, Hcy formation depends on Met intake and on the regulation and function of the Met transmethylation pathway that leads to Hcy formation. The liver is uniquely able to respond to elevated intake or plasma concentration of Met with increased SAM formation because hepatocytes express a liver-specific high Michaelis-Menten constant (Km) isozyme of Met adenosyltransferase that is encoded by the gene MAT1A. Other tissues, as well as the liver, express the low-Km isozyme encoded by the gene MAT2. Although the equilibrium of SAH hydrolase actually favors formation of SAH, the reaction is normally driven forward by rapid removal of the products Hcy and adenosine. Accumulation of SAH can impair transmethylation reactions by allosteric inhibition of methyltransferases.
Fig. 33.2. Methionine metabolism. Numbered reactions are catalyzed by the following enzymes: (1) Met adenosyltransferase; (2) various methyltransferases; (3) adenosylhomocysteine hydrolase; (4) N5-methyl-THF-homocysteine methyltransferase; (5) N5,10-methylene-THF reductase; (6) betaine-homocysteine methyltransferase; (7) cystathionine β-synthase; (8) cystathionine γ-lyase; (9) enzymes involved in polyamine synthesis; and (10) enzymes involved in methylthioadenosine salvage pathway. FAD, flavin adenine dinucleotide; Pi, phosphate; PPi, free pyrophosphate; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced nicotinamide adenine dinucleotide phosphate; PLP, pyridoxal 5′-phosphate; THF, tetrahydrofolate.
The Hcy generated by hydrolysis of SAH has two likely metabolic fates, remethylation and transsulfuration. In remethylation, Hcy acquires a methyl group from N5-methyltetrahydrofolate (N5-methyl-THF) or from betaine to form Met. In transsulfuration, the sulfur is transferred to serine to form Cys, and the remainder of the Hcy molecule is catabolized to α-ketobutyrate and ammonium. Disorders of Hcy remethylation to Met result in Hcy accumulation and reduced regeneration of Met (and hence SAM) by using methyl groups donated directly by N5-methyl-THF or betaine. Remethylation disorders may result from genetic mutations causing a lack of functional Met synthase, a lack of functional coenzyme (methylcobalamin), or a lack of synthesis of the cosubstrate (N5-methyl-THF). Alternatively, a lack of vitamin B12 or folate coenzymes secondary to vitamin deficiency resulting from malabsorption or inadequate intake can also cause a lack of Hcy remethylation. The decrease in SAM levels that accompanies impaired remethylation of Hcy may also lead to decreased transsulfuration and Hcy accumulation because SAM is an important allosteric activator of the transsulfuration enzyme cystathionine β-synthase.
Transsulfuration is the pathway for removal of the Hcy carbon chain as well as for transfer of Met sulfur to serine to synthesize Cys. This pathway is catalyzed by two pyridoxal 5′-phosphate-(PLP)-dependent enzymes: cystathionine β-synthase, which condenses Hcy and serine to form cystathionine; and cystathionine γ-lyase, which hydrolyzes cystathionine to release Cys, α-ketobutyrate, and ammonium. Although all cells are capable of transmethylation and remethylation, the catabolism of Hcy through transsulfuration is restricted to the tissues that express both transsulfuration enzymes. In the rat and mouse, transsulfuration occurs in liver, kidney, pancreas, and intestine (44, 45). Tissues that are not capable of transsulfuration require an exogenous source of Cys and also must export Hcy for further metabolism and removal by other tissues.
As may be predicted, the overexpression of cystathionine β-synthase (on chromosome 21) in children with Down syndrome results in significantly reduced plasma levels of Hcy, Met, SAH, and SAM and in significant increases in plasma cystathionine and Cys (46). In contrast, inborn errors of metabolism that lead to a lack of functional cystathionine β-synthase result in dramatic elevation of tissue and plasma levels of Hcy. Lack of the second enzyme in the transsulfuration pathway, cystathionine γ-lyase, results in accumulation of cystathionine in tissues and the loss of cystathionine in the urine, but no apparent disorder. Nevertheless, a lack of either enzyme impairs the synthesis of Cys from Met (Hcy) sulfur and decreases the supply of Cys to the body.
Met intake provides the substrate for Hcy formation. At typical intakes of SAAs, Hcy formation in men was approximately 19 mmol/day, and Cys formation by transsulfuration of part of this Hcy was approximately 12 mmol/day. In men fed an SAA-free diet, Hcy formation was reduced to 6 mmol/day, and Cys formation was reduced to 2 mmol/day (17, 47). The balance of the Hcy was remethylated back to Met. A major mechanism for regulation of Hcy remethylation versus transsulfuration in response to Met or methyl group availability is the allosteric effects of SAM (48). SAM is both an inhibitor of N5,10-methylene-THF reductase and an activator of cystathionine β-synthase (see also the chapter on folic acid). Hence, when the cellular SAM concentration is low, the synthesis of N5-methyl-THF proceeds uninhibited, and cystathionine synthesis is suppressed, thus favoring Hcy remethylation or Met synthesis. Conversely, when the SAM concentration is high, inhibition of N5-methyl-THF synthesis and stimulation of transsulfuration favor Hcy catabolism and Cys biosynthesis.
Normal adult subjects given a control diet with a betaine supplement had increased rates of Met transmethylation and transsulfuration (49). This finding suggests that an increased dietary supply of methyl groups in the form of choline or betaine may increase Met catabolism by transmethylation and transsulfuration. Presumably, increased remethylation induced by betaine would increase SAM concentration, which would, in turn, result in inhibition of N5-methyl-THF-dependent remethylation and stimulation of cystathionine β-synthase-dependent Hcy catabolism. Thus, a high dietary intake of betaine coupled with a marginal intake of Met could possibly disrupt the normal regulation of Met metabolism and precipitate a Met deficiency state. The importance of betaine or its precursor choline in promoting Hcy remethylation is also supported by the observation that treatment of patients with metabolic syndrome or diabetes mellitus with fibrates resulted in abnormal renal excretion of betaine and a rise in plasma total Hcy (tHcy) (50). Analysis of data obtained in the Framingham Offspring Study (1995 to 1998) that spanned the period before and after folic acid fortification in the United States was analyzed for the association of choline plus betaine intake and plasma tHcy. During the period before supplementation, a higher intake of choline plus betaine was associated with lower concentrations of both fasting tHcy and post-Met-load tHcy, but this association was no longer present in the period after fortification (51).
Cyst(e)ine is said to have a Met-sparing effect by reducing Met catabolism through the transsulfuration pathway, and this process appears to occur with intakes of typical food proteins in which the Met-to-Cys ratio ranges from approximately 1:1 to 2:1 (52). Maximal sparing of Met is approximately 64%, as judged by observations on subjects consuming excess cyst(e)ine and minimal Met (53). The action of supplemental cyst(e)ine when it is added to an SAA-free diet or to a low-Met diet may be explained at least partially by promotion of the incorporation of Met into protein such that less Met is catabolized (54, 55). The action of cyst(e)ine, when it is used to replace part of the dietary Met, thus keeping the total SAA level the same, may be explained by a reduction in the hepatic concentrations of Met and SAM and, hence, less activation and reduced activity of hepatic cystathionine β-synthase. When the Met-to-cyst(e)ine ratio of the diet was increased from 1:0 to 1:1 to 1:3, the ratio of metabolism of Hcy by remethylation versus transsulfuration increased from 0.75 to 1.3 to 1.9 (53). Less catabolism of Hcy by transsulfuration would result in an increase in the recycling of Hcy to Met by using methyl groups generated by the folate coenzyme system (see the chapter on folic acid).
Other mechanisms of regulation of transsulfuration may also play a role in the conversion of Hcy to Cys. Redox regulation of cystathionine β-synthase may provide a means to promote transsulfuration at the expense of remethylation, independently of methylation status, when the body has an increased need for Cys for GSH synthesis. Flux of Hcy through transsulfuration appears to be increased under oxidizing conditions, and this upregulation of transsulfuration has been associated with oxidation of the heme moiety in the N-terminal domain or targeted proteolysis of the C-terminal domain of cystathionine β-synthase (56, 57). In addition, hepatic cystathionine β-synthase gene expression is increased by glucagon and glucocorticoids and decreased by insulin (58, 59). Hormonal regulation of hepatic cystathionine β-synthase expression may serve to conserve Met for protein synthesis in the fed state and to promote catabolism of the Met/Hcy carbon chain to α-ketobutyrate, a gluconeogenic substrate, in the starved state.
Only gold members can continue reading. Log In or Register to continue