Chapter 29 Cutaneous lymphoproliferative diseases and related disorders
Classification of lymphomas
As a result, modern lymphoma classification schemes classify tumors on the basis of constellations of shared morphological, immunophenotypic, genetic, and clinical features, ensuring more relevant disease categories and greater diagnostic consistency. This more systematic approach achieved general acceptance with the publication of the revised European and American lymphoma (REAL) classification in 1994.1 However, although the REAL classification recognized certain cutaneous lymphoproliferative disorders as independent entities, it failed to address the fact that certain lymphomas arising primarily in the skin were biologically distinct from morphologically and phenotypically similar neoplasms occurring in lymph nodes.
The EORTC classification sought to address these deficiencies. This classification scheme was proposed by the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer (EORTC). This was designed for cutaneous lymphomas, but otherwise employed a similar philosophy to that used in the REAL classification, with a greater emphasis on the unique clinical features of some primary cutaneous lymphomas.2 It was originally based on the data derived from 626 patients in the records of the Dutch Registry for Cutaneous Lymphoma, and later validated in several large studies with follow-up data from > 1300 patients.2–5 This classification subdivides primary cutaneous lesions into indolent, aggressive, and provisional categories based on behavior as determined by clinical experience (Table 29.1). This was a major step forward because it provided a precise definition for a number of primary cutaneous lymphomas and recognized differences in biological behavior (and thus potential treatment requirements) between nodal and cutaneous primary disease. For example, CD30-positive (ALK-negative) large T-cell lymphomas arising in lymph nodes are high-grade tumors, with a mortality of up to 63% whereas morphologically and immunophenotypically identical tumors arising primarily in the skin are clinically low grade, and have a mortality of less than 5%.6,7
Table 29.1 EORTC classification of primary cutaneous lymphomas
Modified from: Willemze, R., et al. (1997) Blood, 90, 354–371.
| Primary CTCL | Primary CBCL |
|---|---|
| Indolent | |
| Mycosis fungoides | Follicle center cell lymphoma |
| Mycosis fungoides + follicular mucinosis | Immunocytoma(marginal zone B-cell lymphoma) |
| CD30+ large cell CTCL | |
| Lymphomatoid papulosis | |
| Intermediate | |
| Large B-cell lymphoma of the leg | |
| Aggressive | |
| Sézary’s syndrome | |
| CD30− large cell CTCL | |
| Provisional | |
| Granulomatous slack skin | Intravascular large B-cell lymphoma |
| CTCL, pleomorphic small/medium-sized | Plasmacytoma |
| Subcutaneous panniculitis-like T-cell lymphoma |
Abbreviations: CTCL, cutaneous T-cell lymphoma; CBCL, cutaneous B-cell lymphoma
Many of the advances made in the EORTC classification were not recognized in the update of the REAL classification published jointly with the WHO in 2001.8 This generated considerable controversy, particularly with respect to B-cell neoplasms. Arguments used against the EORTC approach included complaints of too broad a definition of primary cutaneous follicle center cell lymphoma, and the appropriateness of designating large B-cell lymphoma of the leg as a separate entity.9,10 Dissention also reigned over use of the term primary cutaneous ‘immunocytoma’ as opposed to marginal zone lymphoma, immunocytoma at that time being inclusive of lymphoplasmacytic lymphoma, an entity distinct from marginal zone lymphoma.
These differences have been resolved following meetings of members of both classification systems in Lyon and Zurich in 2003 and 2004. This resulted in a consensus WHO-EORTC classification first published in 2005.11,12 The entities delineated in this system have since been incorporated, with refinements of some diagnostic criteria and the addition of new entities, into the updated WHO classification of tumors of the hematopoietic and lymphoid tissue published in 2008.13 This version of the WHO classification will be used in this chapter. The main lymphoma subtypes that primarily affect the skin are listed along with their expectant clinical behavior in Table 29.2.
Table 29.2 Lymphoma subtypes and behavior as defined in WHO-EORTC classification of cutaneous lymphoma
| WHO-EORTC subtype | Behavior |
|---|---|
| Cutaneous T-cell lymphoma | |
| Mycosis fungoides | Indolent |
| MF variants | |
| Folliculotropic MF | Indolent |
| Pagetoid reticulosis | Indolent |
| Granulomatous slack skin | Indolent |
| Sézary’s syndrome | Aggressive |
| Adult T-cell leukemia/lymphoma | Aggressive |
| Primary cutaneous CD30+ lymphoproliferative disorders | |
| Primary cutaneous anaplastic large cell lymphoma | Indolent |
| Lymphomatoid papulosis | Indolent |
| Subcutaneous panniculitis-like T-cell lymphoma | Indolent |
| Extranodal NK/T-cell lymphoma, nasal type | Aggressive |
| Primary cutaneous T-cell lymphoma, unspecified | |
| Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma* | Aggressive |
| WHO-EORTC subtype | Behavior |
| Cutaneous γ/δ T-cell lymphoma* | Aggressive |
| Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma* | Indolent |
| Cutaneous B-cell lymphoma | |
| Primary cutaneous marginal zone lymphoma | Indolent |
| Primary cutaneous follicle center lymphoma | Indolent |
| Primary cutaneous large B-cell lymphoma, leg type | Intermediate |
| Primary cutaneous large B-cell lymphoma, other | Intermediate |
| Primary cutaneous intravascular large B-cell lymphoma | Intermediate |
| Precursor hematological neoplasm | |
| CD4+/CD56+ hematodermic neoplasm (blastic plasmacytoid dendritic cell neoplasm) | Aggressive |
Abbreviations: MF, mycosis fungoides
Modified from: Willemze, R., et al. (2005). Blood, 105, 3768–3785.
T-CELL LYMPHOMAS
Mycosis fungoides
Clinical features
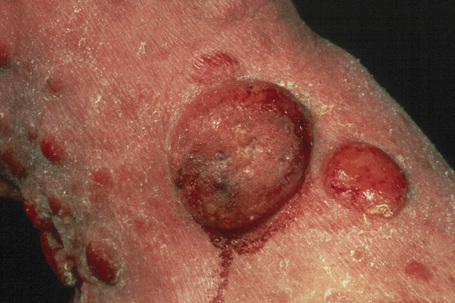
Mycosis fungoides (MF) (Gr. mykes, fungus; L. fungus + Gr. eidos, form), although rare, represents the commonest form of primary cutaneous T-cell lymphoma.1–7 Alibert named it, in 1806, after the mushroom-like tumors that develop in the terminal stages of the illness (Fig. 29.1). The annual incidence in the USA varies from 0.36 to 0.46 cases per 105 of the population8–10 with approximately 1000 new cases diagnosed per year.1,11 The incidence in Europe is somewhat less.12 There is predilection for males (2:1). It is more common in blacks (2:1) and less common in Asians and Hispanics.2,8,11,13 Any age group may be involved but there is a higher incidence in the fourth to sixth decades. MF in children is discussed separately (see below).
The course and outcome is unpredictable, ranging from a protracted, persistent, relatively benign illness through to a widespread malignancy with high morbidity and mortality.3
Rarer presentations include bullous, follicular, hypopigmented, verrucous/hyperkeratotic, pustular, lichenoid papular, palmoplantar psoriasiform, granulomatous, and acanthosis nigricans-like variants. These are clinically unusual cases that run a similar course to that of classic MF, and are not considered separate entities. In contrast, folliculotropic mycosis fungoides, pagetoid reticulosis, and granulomatous slack skin disease, have distinct clinicopathological features and are recognized as biologically distinct variants of MF in the new WHO classification.14
Classic MF is traditionally divided into patch, plaque, and tumor stages.2 This is, however, a somewhat arbitrary classification because all stages may be present simultaneously in one individual while other patients never progress beyond the patch stage.15,16 In addition, patch stage lesions obviously merge with plaques.
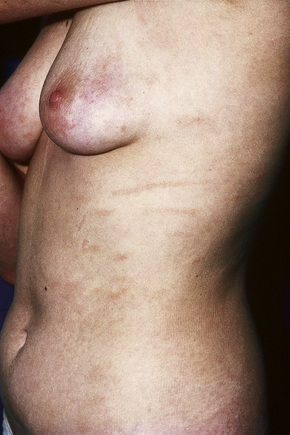
The early erythematous lesions are irregular, asymmetrical, slightly scaly, variably sized pink or red patches (Fig. 29.2). Many lesions show signs of atrophy and in some lighting conditions they appear smooth and shiny. While lesions are more commonly present on the trunk, limb girdles, breasts, and flexures, they can be more widespread (Figs 29.3, 29.4).
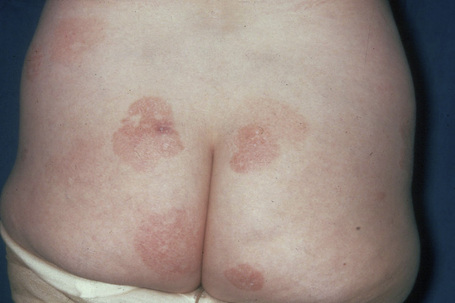
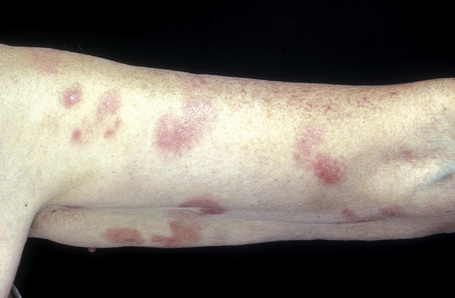
Fig. 29.3 Mycosis fungoides: multiple patches are present on this patient’s arm.
By courtesy of the Institute of Dermatology, London, UK.
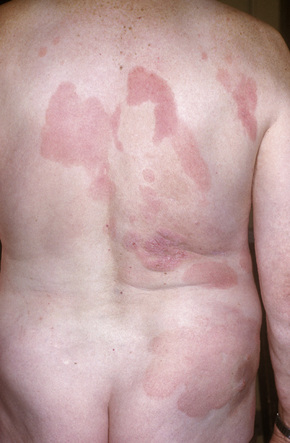
Fig. 29.4 Mycosis fungoides: lesions sometimes have a generalized distribution.
By courtesy of the Institute of Dermatology, London, UK.
The clinical differential diagnosis includes discoid, atopic or contact allergic dermatitis, psoriasis and, in particular, chronic superficial scaly dermatitis (small plaque parapsoriasis).17–22 Patients, usually middle-aged, present with erythematous scaly persistent patches, showing predilection for the limbs and trunk, that are sometimes likened to cigarette paper.20,23 While the lesions may be round or oval, they often have a finger-like appearance – hence the alternative designation digitate dermatosis (Fig. 29.5). The patches tend to be uniform in size, shape, and color, contrasting vividly with the great variability of those of mycosis fungoides.
Adverse drug reactions may also mimic mycosis fungoides. Patients can present with multiple infiltrated plaques or erythroderma that are histologically indistinguishable from mycosis fungoides.22 Drugs which have been particularly implicated are the anticonvulsants (including phenytoin, barbiturates, carbamazepine), cardiac drugs such as atenolol and angiotensin-converting enzyme (ACE) inhibitors, antihistamines, ciclosporin, and allopurinol.22,24–26
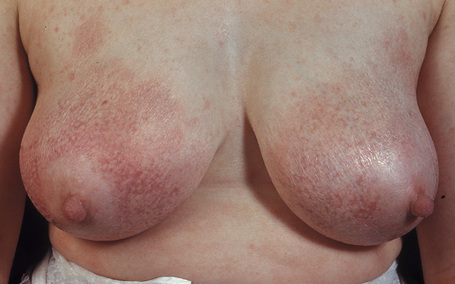
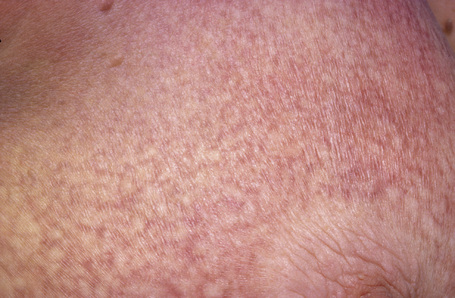
Patients sometimes develop foci of poikiloderma within a more typical background. In others, the entire eruption may be poikilodermatous – so-called poikiloderma atrophicans vasculare (Gr. poikilos, spotted, mottled, varied) (large plaque parapsoriasis).17 They present with small numbers of large plaques showing a predilection for the breasts, buttocks, hips, abdomen, and major flexures (Fig. 29.6). Individual features of the plaques include atrophy, telangiectases, and variable hypo- and hyperpigmentation with erythema (Fig. 29.7). The appearances have been likened to those of chronic radiation damage. Further progression may be similar to that of classic mycosis fungoides, although it appears that fewer patients develop tumor-stage mycosis fungoides.27
Further progression of patch stage disease leads to the development of an increased number of indurated plaques (Figs 29.8, 29.9). The lesions can be quite bizarre and, not uncommonly, due to central regression, they have an annular or serpiginous appearance (Fig. 29.10). Plaques are sometimes extremely hyperkeratotic, particularly on the palms and soles (see mycosis fungoides palmaris et plantaris).
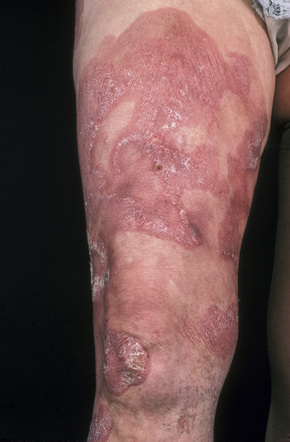
Fig. 29.8 Mycosis fungoides: large erythematous plaques with scaling.
By courtesy of the Institute of Dermatology, London, UK.
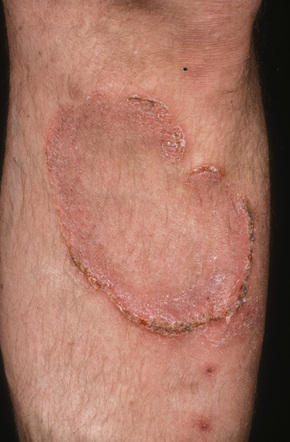
Fig. 29.9 Mycosis fungoides: high-power view.
By courtesy of the Institute of Dermatology, London, UK.
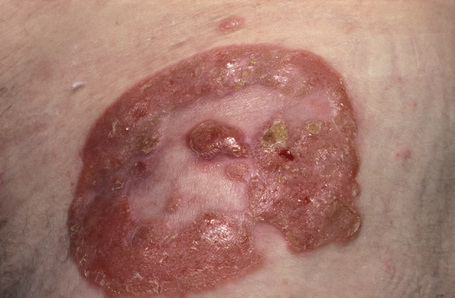
Fig. 29.10 Mycosis fungoides: close-up view of an annular lesion.
By courtesy of the late N.P. Smith, MD, Institute of Dermatology, London, UK.
A small proportion of patients develop tumors (Figs 29.11–29.13). Sometimes ulcerative lesions are seen (Fig.29.14). The face, scalp, and intertriginous areas are particularly affected.28
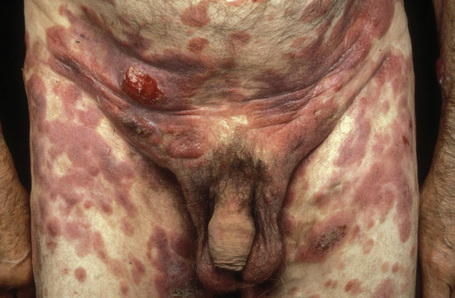
Fig. 29.11 Mycosis fungoides: multiple tumors have arisen against a background of plaque stage disease.
By courtesy of the Institute of Dermatology, London, UK.
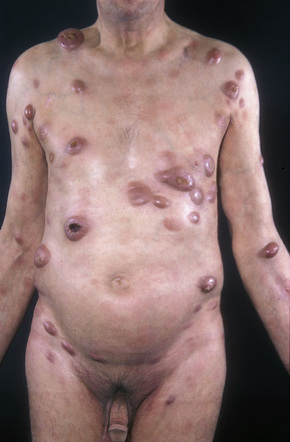
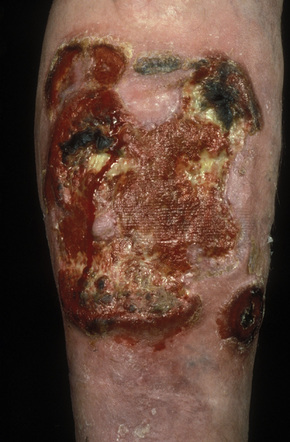
Fig. 29.13 Mycosis fungoides: this patient shows advanced disease with a fungating tumor on the knee.
By courtesy of the Institute of Dermatology, London, UK.
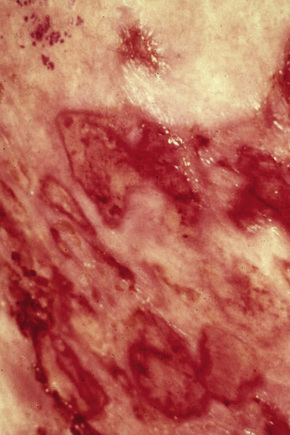
Fig. 29.14 Mycosis fungoides: ulcerative lesions, as seen in this patient, are a rare manifestation.
By courtesy of A. du Vivier, MD, King’s College Hospital, London, UK.
Clinical staging relates to the extent of disease and the presence or absence of cutaneous tumor nodules and erythroderma. The International Society for Cutaneous Lymphomas (ISCL) and the EORTC recently issued a new proposal for staging and classification of mycosis fungoides and Sézary’s syndrome.29 This proposal takes account of advances in molecular biology, immunohistochemistry, and imaging, as well as new data on prognostic variables. It assesses the extent and nature of the skin lesions present, extent of nodal involvement, visceral involvement, and the presence and degree of any peripheral blood involvement (Table 29.3). Combinations of these parameters (stage) can be used to determine prognosis and are essential for determining treatment.29,30 Outcome in mycosis fungoides is varied. When only patches and plaques are present at presentation, and cover 10% of body surface area (T1), survival is no different from age-matched controls.16,31–33 Patients with patches and plaques covering > 10% body surface area (T2) have a median survival of 10–12 years and a 25% risk of progression, whilst the median survival for patients with tumors (T3) or erythroderma (T4) is only 4–5 years.32,33 Visceral involvement carries a very poor prognosis, with median survivals of only 1–2 years.30,33 Lymph node enlargement is common and does not necessarily indicate pathological involvement. Its impact on prognosis is often overshadowed by the extent of skin lesions. The presence of abnormal circulating cells and the tumor burden in the peripheral blood are also important parameters to assess as they are of independent prognostic significance.32,34,35 The patient should therefore be thoroughly investigated to assess the extent of skin lesions and to determine whether there is nodal, visceral or hematological spread.
Table 29.3 ISCL/EORTC revision to clinical and pathological staging of mycosis fungoides
| Stage | Description | TNMB |
|---|---|---|
| IA | Limited patches, papules and/or plaques covering < 10% skin surface area (T1); no evidence of nodal (No), visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1) | T1N0M0B0,1 |
| B | Generalized patches, papules and/or plaques covering ≥ 10% skin surface area (T2); no evidence of nodal (No), visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1) | T2N0M0B0,1 |
| II | Any extent of patches, papules and/or plaques (T! 0r T2); clinically abnormal nodes showing DL (N1) or early involvement by MF (N2); no evidence of visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1) | T1,2N1,2M0B0,1 |
| IIB | One or more tumors (≥1.5 cm diameter) (T3); up to early lymph node involvement (N2); no evidence of visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1) | T3N0-2M0B0,1 |
| III | Erythema covering ≥ 80% skin surface area; up to early lymph node involvement (N2); no evidence of visceral (M0) or significant blood involvement (B0), or low blood tumor burden (B1) | T4N0-2M0B0,1 |
| IIIA | Stage III with no significant blood involvement | T4N0-2M0B0 |
| IIIB | Stage III with low blood tumor burden | T4N0-2M0B1 |
| IVA1 | Any degree of skin involvement; up to early lymph node involvement; no evidence of visceral spread; high blood tumor burden (B2: > 1000 Sezary cells/ml with positive clone) | T1-4N0-2M0B2 |
| IVA2 | Any degree of skin involvement; partial or complete effacement of node by MF (N3); no visceral involvement; any degree of blood involvement | T1-4N3M0B0-2 |
| IVB | Any degree of skin involvement with visceral involvement, irrespective of nodal or blood involvement | T1-4N0-3M1B0-2 |
Modified from Olsen, E., et al. (2007) Blood, 110, 1713–1722.
Raised levels of lactate dehydrogenase, soluble interleukin 2 receptor, erythrocyte sedimentation rate, and raised blood eosinophil count are other markers of poor prognosis.36–42 Transformation to large cell lymphoma also relates to adverse outcome. Conversely, other histological parameters that are associated with improved results include increased numbers of reactive CD8-positive T cells, increased numbers of FoxP3-positive T-regulatory cells and increased numbers of CD1a-positive dendritic cells.43–45 In addition, the presence of identical TCR gene rearrangements in multiple biopsy specimens at the time of diagnosis has been shown to predict for disease progression.46
Infection is the major cause of death, with Staphylococcus aureus, Enterobacteriaceae and Pseudomonas aeruginosa being the most frequent pathogens.1,2,47–49 Beta-hemolytic streptococcus, herpes simplex, and varicella-zoster infections are also of importance.48
Patients with mycosis fungoides also have an increased risk of epithelial tumors, including carcinoma of the lung and colon, and B-cell non-Hodgkin’s lymphoma.50–52
Pathogenesis and histological features
There is no longer any doubt that the condition represents a neoplastic process from the beginning. Monoclonal T-cell receptor (TCR) gene rearrangements have been identified using standard molecular techniques such as Southern blot (TCRβ) and polymerase chain reaction (PCR) (TCRγ and TCRβ) analyses in up to 90% of mycosis fungoides patients.53–60 While monoclonal lymphoid populations have been described in apparently non-neoplastic conditions, such as pityriasis lichenoides acuta, lichen aureus, lichen planus, pigmented purpuric dermatosis, allergic contact dermatitis, and drug reactions, the presence of a clonal TCR gene rearrangement is highly suggestive of a lymphomatous process, particularly when the same clone is found in more than one lesion or in one patient over time.58–72 Recurrent chromosomal abnormalities have been found in some cases including loss of chromosomes 1p, 17p, 10q/10, 13q, and 19, as well as gains of 4/4q, 17q/17, and 18.73
As with many malignancies, the etiology and pathogenesis of mycosis fungoides is likely to be multifactorial. Specific causative agents and precise pathogenetics are unknown.74
There is little evidence of genetic predisposition to mycosis fungoides. Familial occurrence is exceptional.75,76 A number of studies have linked various human leukocyte antigen (HLA) types with mycosis fungoides. In the UK, HLA-B8 and HLA-AW19 have been found with increased frequency.77 This finding was not confirmed in a US study.78 Subsequently, an association with the antigens HLA-DR5 (DRB1*11) and DQB1*03 was demonstrated in North American Caucasians.79,80 In studies from Israel, no association with HLA class I alleles was detected.81 There was, however, a statistically significant increased incidence of HLA-DQB1*03 and HLA-DRB1*11 in Ashkenazi Jews.76 Whether these findings indicate an increased susceptibility to or a role in the pathogenesis of mycosis fungoides is uncertain.
Early epidemiological studies provided conflicting results on the potential association of occupational factors, including exposure to metals, plastics, cutting oils, and solvents.10,82 More recent case-control studies have, however, identified an increased risk of mycosis fungoides in certain occupations such as glass, pottery, and ceramic workers, and suggest that exposure to aromatic and/or halogenated hydrocarbons or pesticides is implicated in its pathogenesis.83–85
It is thought that mycosis fungoides most probably develops as a consequence of chronic antigenic stimulation and it has long been postulated that infective agents are likely to play a pathogenetic role.3,74,86 A wide range of potential agents, particularly viruses, have been investigated including HTLV-I, HTLV-II, Epstein-Barr virus (EBV), and cytomegalovirus. However, the results of various studies looking for evidence of these agents in MF have produced conflicting or inconclusive results.6,74
Recently, attention has focused on a possible association between human herpes virus 8 (HHV8) and MF. Some studies have not detected viral-specific DNA in lesional tissues of MF patients by PCR.87,88 However, a recent investigation detected HHV8-specific DNA by PCR in 70% of biopsies of affected patients, but found no evidence of latency-associated nuclear antigen using immunohistochemistry, suggesting low levels of the virus in many cases.89 HHV6 and HHV7 have been found in only a minority of cases.87,90
Other infective agents postulated to have an etiological role include Staphylococcus aureus, persistent chlamydia infection and Borrelia burgdorferi.83,91–94 However, there is no strong evidence in favor of these agents playing a direct role in MF pathogenesis.74 Nevertheless, it remains possible that various infectious agents are important initiators of the chronic lymphoproliferation from which lymphoma eventually develops.
The neoplastic cells express cytokine receptors CCR4 and CCR10.95,96 CCL17 and CCL22 are ligands for CCR4 and are present in high levels in skin lesions of affected patients.95,97 CCL17 is also increased in the serum of patients with the disease and Sézary’s syndrome, as is CCL27, the ligand for CCR10.74 Interaction of CCR4 and CCR10 with their respective ligands not only influences T-cell migratory properties, but promotes their survival via downstream activation of antiapoptotic pathways such as phosphatidylinositol-3-kinase and Akt.74 Such chemokine interactions are therefore likely to be important in initiation and perpetuation of the skin lesions and may explain the marked propensity for the malignant cells to localize in the epidermis.97
MF cells appear to be in a persistent state of activation. They express cell surface adhesion molecules such as CD45RO and IL2R.98,99 There is evidence of constitutive activation of several intracellular signaling proteins in the JAK-STAT family of molecules, similar to that witnessed following stimulation of normal T cells via the IL-2 receptor.100–103 This is likely to affect regulation of various aspects of cell survival and proliferation. Other factors may also contribute towards defects in normal apoptotic pathways in MF cells. For example, mutations of the Fas gene or abnormal splice variants of its transcript have been documented in MF and may protect against Fas-mediated stimulation of apoptosis.104,105 Also, gene expression profiling studies have recently generated a genetic signature specific for MF. This is characterized by multiple genes involved in the TNF signaling pathway, including several inhibitors of apoptosis.106 Abnormalities in Fas and TNF signaling pathways may be responsible for the constitutive activation of the NF-kappa B pathway noted in MF, further supporting a role for resistance to apoptosis in its pathogenesis.107
Alterations in a number of tumor suppressor and apoptosis-related genes have also been detected. Silencing of p15, p16, Nav3, PTEN, and p53 due to mutations, promoter hypermethylation or allelic loss have all been implicated in the progression from plaque to tumor stage.104,108–111
Tumor cells in MF also seem equipped to evade the host immune system. Cytotoxic T cells can induce apoptosis via engagement of Fas, expressed on the surface of the target cell, by Fas-ligand (FasL). The aforementioned defects in Fas-ligand signaling in MF cells is therefore likely to protect against tumor-specific cytotoxic T-cell-mediated immunity.97,104,105 Furthermore, the malignant cells in MF may express FasL, giving them the potential to eliminate tumor-specific CD8-positive cytotoxic T cells.112 They also show down- regulation of Th1-associated genes, and express cytokines such as interleukin-4 (IL-4), IL-5 and IL-18, most in keeping with Th2-type T cells.113–115 This Th2 bias, together with production of other immunomodulatory cytokines such as IL-10, transforming growth factor-β, and soluble IL-2R may also contribute to suppressing the host antitumor immune response and partly account for the increased risk of infections and second malignancies.116–119 Copy number gains and amplifications of JUN B, and JUN B overexpression, have been documented in MF and may contribute to the Th2 bias, as JUNB promotes IL4 mediated TH2 lymphocyte differentiation.120,121
The lymphocytes of MF are mature T cells that have transited through the lymph node and undergone antigenic stimulation. They show a particular tendency to colonize the epidermis (epidermotropism), although this is more evident in the patch and plaque forms than in tumor-stage disease. To a lesser extent in early lesions, and more obviously in later stages, the infiltrate contains large cells with highly irregular, convoluted or cerebriform nuclei, known as Sézary or mycosis cells (Fig. 29.15). Assessing their significance must be tempered with caution because identical cells (albeit in small numbers) are sometimes seen in the dermal infiltrate of a variety of dermatoses. Their presence must obviously be considered in the context of the accompanying histological features and, in particular, in the light of the clinical information. Electron microscopically, mycosis/Sézary cells are characterized by a multilobed hyperconvoluted nucleus with conspicuous peripheral chromatin margination (Fig. 29.16).
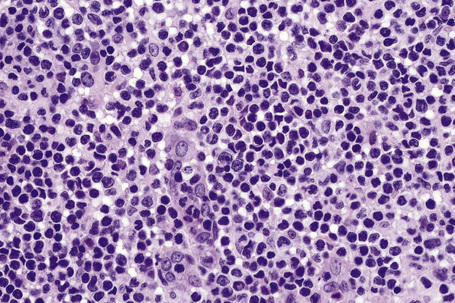
Fig. 29.15 Mycosis fungoides: the nuclei of the mycosis cells are hyperchromatic and highly irregular.
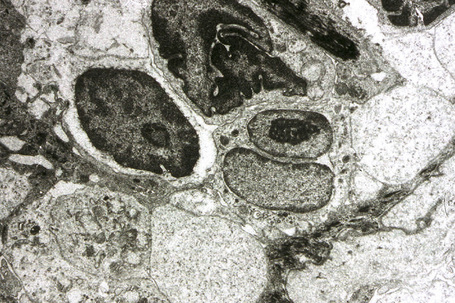
Epidermotropism is therefore the histological hallmark of MF.122–131 It is related to the stage of the disease and the degree of differentiation of the lymphocytes. With the development of tumor stage MF, large transformed cells are conspicuous and epidermotropism is commonly lost. Epidermotropism as seen in cutaneous T-cell lymphoma differs from exocytosis of lymphocytes as seen in dermatitis/eczema in that there is usually no or only very mild spongiosis and vesiculation is not usually a feature. The presence of atypical lymphoid cells in an intraepidermal vesicle (the Pautrier microabscess) is typical, although not diagnostic, of mycosis fungoides (Fig. 29.17). Similar features may be seen, though less often, in Sézary’s syndrome, adult T-cell leukemia/lymphoma (ATLL), actinic reticuloid and drug-induced T-cell pseudolymphomas.
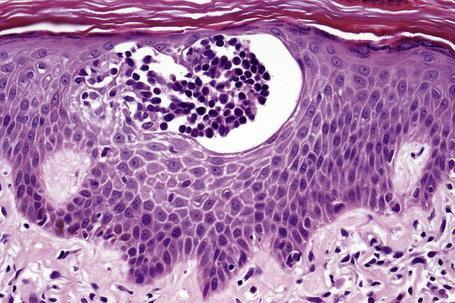
Fig. 29.17 Mycosis fungoides: a typical Pautrier microabscess composed of hyperchromatic atypical lymphocytes.
Patch stage mycosis fungoides
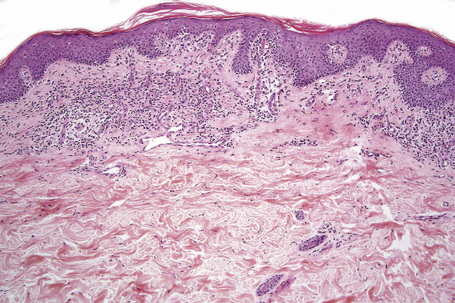
The histopathological features of the early patch stage of MF are usually subtle and easily overlooked (Fig. 29.18). Multiple biopsies are commonly necessary to reach a diagnosis. The epidermis may be of normal thickness, slightly acanthotic or less commonly atrophic, and often there is mild hyperkeratosis with focal parakeratosis. Basal cell hydropic degeneration is sometimes noted. Within the epidermis, there are characteristically small numbers of atypical irregular lymphoid cells, each surrounded by a clear halo, although in very early lesions they may sometimes be absent (Fig. 25.19). Occasionally, small numbers of larger typical cells may also be evident. Palisading of lymphocytes along the basal layer of the epidermis is a diagnostic pointer. Individual necrotic keratinocytes are seen in some cases. A lymphohistiocytic infiltrate surrounds the vessels of the superficial vascular plexus and extends into the papillary dermis (Fig. 29.20). Eosinophils and plasma cells are present in small numbers or absent.129,132 Red cell extravasation is sometimes evident. Pigmentary incontinence is common. The connective tissue of the papillary dermis is occasionally increased with coarsening of the collagen bundles (although this is more typical of plaque stage disease).129 In the more advanced patch stage, increasing numbers of atypical cells may make the diagnosis more obvious, although frank Pautrier microabscesses are uncommon (Figs 29.21, 29.22).
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


