(Table 72-9) Disorders of hypopigmentation are often classified as either diffuse or localized. The classic example of diffuse hypopigmentation is oculocutaneous albinism (OCA). The most common forms are due to mutations in the tyrosinase gene (type I) or the P gene (type II); patients with type IA OCA have a total lack of enzyme activity. At birth, different forms of OCA can appear similar—white hair, gray-blue eyes, and pink-white skin. However, the patients with no tyrosinase activity maintain this phenotype, whereas those with decreased activity will acquire some pigmentation of the eyes, hair, and skin as they age. The degree of pigment formation is also a function of racial background, and the pigmentary dilution is more readily apparent when patients are compared to their first-degree relatives. The ocular findings in OCA correlate with the degree of hypopigmentation and include decreased visual acuity, nystagmus, photophobia, strabismus, and a lack of normal binocular vision.
CAUSES OF HYPOPIGMENTATION |
aAbsence of melanocytes in areas of leukoderma. bNormal number of melanocytes. cPlatelet storage defect and restrictive lung disease secondary to deposits of ceroid-like material or immunodeficiency; due to mutations in β subunit of adaptor protein 3 as well as subunits of biogenesis of lysosome-related organelles complex (BLOC)-1, -2, and -3. dGiant lysosomal granules and recurrent infections. eMinority of patients in a nonreferral setting have systemic abnormalities (musculoskeletal, central nervous system, ocular).
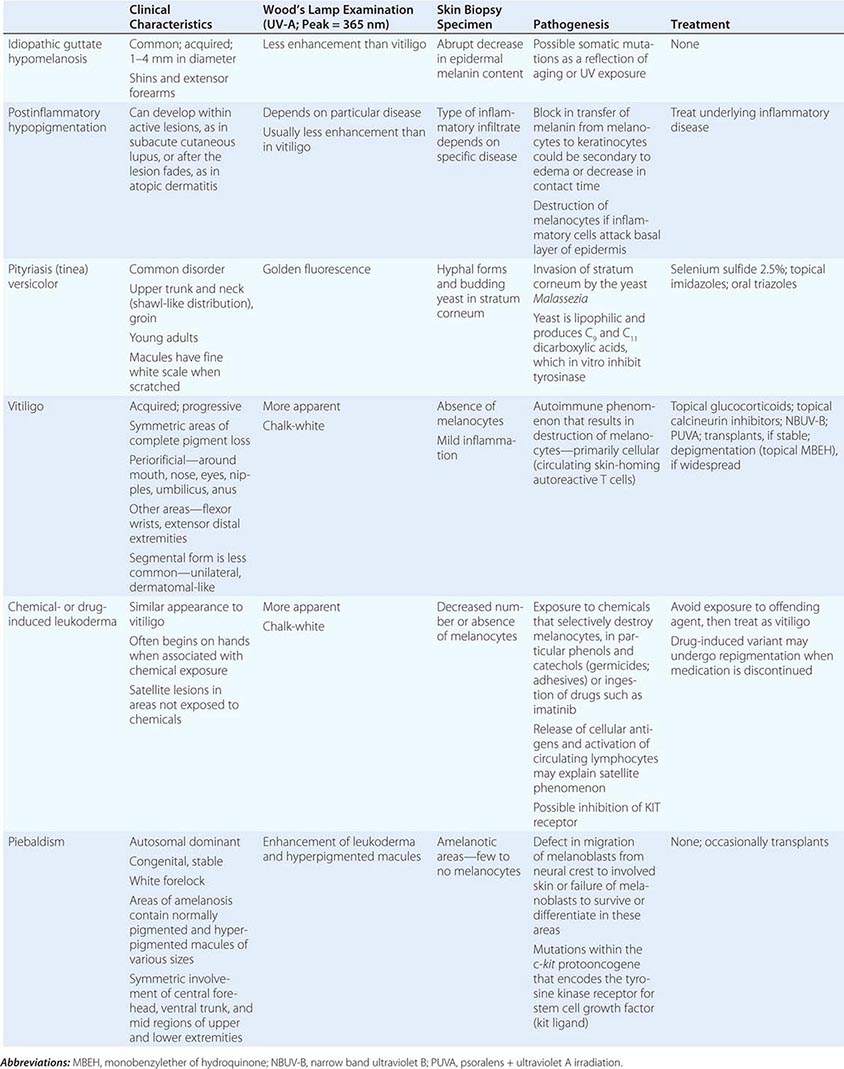
The differential diagnosis of localized hypomelanosis includes the following primary cutaneous disorders: idiopathic guttate hypomelanosis, postinflammatory hypopigmentation, tinea (pityriasis) versicolor, vitiligo, chemical- or drug-induced leukoderma, nevus depigmentosus (see below), and piebaldism (Table 72-10). In this group of diseases, the areas of involvement are macules or patches with a decrease or absence of pigmentation. Patients with vitiligo also have an increased incidence of several autoimmune disorders, including Hashimoto’s thyroiditis, Graves’ disease, pernicious anemia, Addison’s disease, uveitis, alopecia areata, chronic mucocutaneous candidiasis, and the autoimmune polyendocrine syndromes (types I and II). Diseases of the thyroid gland are the most frequently associated disorders, occurring in up to 30% of patients with vitiligo. Circulating autoantibodies are often found, and the most common ones are antithyroglobulin, antimicrosomal, and antithyroid-stimulating hormone receptor antibodies.
HYPOPIGMENTATION (PRIMARY CUTANEOUS DISORDERS, LOCALIZED) |
There are four systemic diseases that should be considered in a patient with skin findings suggestive of vitiligo—Vogt-Koyanagi-Harada syndrome, systemic sclerosis, onchocerciasis, and melanoma-associated leukoderma. A history of aseptic meningitis, nontraumatic uveitis, tinnitus, hearing loss, and/or dysacousia points to the diagnosis of the Vogt-Koyanagi-Harada syndrome. In these patients, the face and scalp are the most common locations of pigment loss. The vitiligo-like leukoderma seen in patients with systemic sclerosis has a clinical resemblance to idiopathic vitiligo that has begun to repigment as a result of treatment; that is, perifollicular macules of normal pigmentation are seen within areas of depigmentation. The basis of this leukoderma is unknown; there is no evidence of inflammation in areas of involvement, but it can resolve if the underlying connective tissue disease becomes inactive. In contrast to idiopathic vitiligo, melanoma-associated leukoderma often begins on the trunk, and its appearance, if spontaneous, should prompt a search for metastatic disease. It is also seen in patients undergoing immunotherapy for melanoma, including ipilimumab, with cytotoxic T lymphocytes presumably recognizing cell surface antigens common to melanoma cells and melanocytes, and is associated with a greater likelihood of a clinical response.
There are two systemic disorders (neurocristopathies) that may have the cutaneous findings of piebaldism (Table 72-9). They are Shah-Waardenburg syndrome and Waardenburg syndrome. A possible explanation for both disorders is an abnormal embryonic migration or survival of two neural crest–derived elements, one of them being melanocytes and the other myenteric ganglion cells (leading to Hirschsprung disease in Shah-Waardenburg syndrome) or auditory nerve cells (Waardenburg syndrome). The latter syndrome is characterized by congenital sensorineural hearing loss, dystopia canthorum (lateral displacement of the inner canthi but normal interpupillary distance), heterochromic irises, and a broad nasal root, in addition to the piebaldism. The facial dysmorphism can be explained by the neural crest origin of the connective tissues of the head and neck. Patients with Waardenburg syndrome have been shown to have mutations in four genes, including PAX-3 and MITF, all of which encode DNA-binding proteins, whereas patients with Hirschsprung disease plus white spotting have mutations in one of three genes—endothelin 3, endothelin B receptor, and SOX-10.
In tuberous sclerosis, the earliest cutaneous sign is macular hypomelanosis, referred to as an ash leaf spot. These lesions are often present at birth and are usually multiple; however, detection may require Wood’s lamp examination, especially in fair-skinned individuals. The pigment within them is reduced, but not absent. The average size is 1–3 cm, and the common shapes are polygonal and lance-ovate. Examination of the patient for additional cutaneous signs such as multiple angiofibromas of the face (adenoma sebaceum), ungual and gingival fibromas, fibrous plaques of the forehead, and connective tissue nevi (shagreen patches) is recommended. It is important to remember that an ash leaf spot on the scalp will result in a circumscribed patch of lightly pigmented hair. Internal manifestations include seizures, mental retardation, central nervous system (CNS) and retinal hamartomas, pulmonary lymphangioleiomyomatosis (women), renal angiomyolipomas, and cardiac rhabdomyomas. The latter can be detected in up to 60% of children (<18 years) with tuberous sclerosis by echocardiography.
Nevus depigmentosus is a stable, well-circumscribed hypomelanosis that is present at birth. There is usually a single oval or rectangular lesion, but when there are multiple lesions, the possibility of tuberous sclerosis needs to be considered. In linear nevoid hypopigmentation, a term that is replacing hypomelanosis of Ito and segmental or systematized nevus depigmentosus, streaks and swirls of hypopigmentation are observed. Up to a third of patients in a tertiary care setting had associated abnormalities involving the musculoskeletal system (asymmetry), the CNS (seizures and mental retardation), and the eyes (strabismus and hypertelorism). Chromosomal mosaicism has been detected in these patients, lending support to the hypothesis that the cutaneous pattern is the result of the migration of two clones of primordial melanocytes, each with a different pigment potential.
Localized areas of decreased pigmentation are commonly seen as a result of cutaneous inflammation (Table 72-10) and have been observed in the skin overlying active lesions of sarcoidosis (see “Papulonodular Skin Lesions,” below) as well as in CTCL. Cutaneous infections also present as disorders of hypopigmentation, and in tuberculoid leprosy, there are a few asymmetric patches of hypomelanosis that have associated anesthesia, anhidrosis, and alopecia. Biopsy specimens of the palpable border show dermal granulomas that contain rare, if any, Mycobacterium leprae organisms.
HYPERPIGMENTATION
(Table 72-11) Disorders of hyperpigmentation are also divided into two groups—localized and diffuse. The localized forms are due to an epidermal alteration, a proliferation of melanocytes, or an increase in pigment production. Both seborrheic keratoses and acanthosis nigricans belong to the first group. Seborrheic keratoses are common lesions, but in one rare clinical setting, they are a sign of systemic disease, and that setting is the sudden appearance of multiple lesions, often with an inflammatory base and in association with acrochordons (skin tags) and acanthosis nigricans. This is termed the sign of Leser-Trélat and alerts the clinician to search for an internal malignancy. Acanthosis nigricans can also be a reflection of an internal malignancy, most commonly of the gastrointestinal tract, and it appears as velvety hyperpigmentation, primarily in flexural areas. However, in the majority of patients, acanthosis nigricans is associated with obesity and insulin resistance, although it may be a reflection of an endocrinopathy such as acromegaly, Cushing’s syndrome, polycystic ovary syndrome, or insulin-resistant diabetes mellitus (type A, type B, and lipodystrophic forms).
CAUSES OF HYPERPIGMENTATION |
aAlso lentigines. bPolyostotic fibrous dysplasia. cSee also “Papulonodular Skin Lesions.” dLate 1980s.
Abbreviations: LAMB, lentigines, atrial myxomas, mucocutaneous myxomas, and blue nevi; LEOPARD, lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis and subaortic valvular stenosis, abnormal genitalia, retardation of growth, and deafness (sensorineural); NAME, nevi, atrial myxoma, myxoid neurofibroma, and ephelides (freckles); POEMS, polyneuropathy, organomegaly, endocrinopathies, M-protein, and skin changes.
A proliferation of melanocytes results in the following pigmented lesions: lentigo, melanocytic nevus, and melanoma (Chap. 105). In an adult, the majority of lentigines are related to sun exposure, which explains their distribution. However, in the Peutz-Jeghers and LEOPARD (lentigines; ECG abnormalities, primarily conduction defects; ocular hypertelorism; pulmonary stenosis and subaortic valvular stenosis; abnormal genitalia [cryptorchidism, hypospadias]; retardation of growth; and deafness [sensorineural]) syndromes, lentigines do serve as a clue to systemic disease. In LEOPARD syndrome, hundreds of lentigines develop during childhood and are scattered over the entire surface of the body. The lentigines in patients with Peutz-Jeghers syndrome are located primarily around the nose and mouth, on the hands and feet, and within the oral cavity. While the pigmented macules on the face may fade with age, the oral lesions persist. However, similar intraoral lesions are also seen in Addison’s disease, in Laugier-Hunziker syndrome (no internal manifestations), and as a normal finding in darkly pigmented individuals. Patients with this autosomal dominant syndrome (due to mutations in a novel serine threonine kinase gene) have multiple benign polyps of the gastrointestinal tract, testicular or ovarian tumors, and an increased risk of developing gastrointestinal (primarily colon) and pancreatic cancers.
In the Carney complex, numerous lentigines are also seen, but they are in association with cardiac myxomas. This autosomal dominant disorder is also known as the LAMB (lentigines, atrial myxomas, mucocutaneous myxomas, and blue nevi) syndrome or NAME (nevi, atrial myxoma, myxoid neurofibroma, and ephelides [freckles]) syndrome. These patients can also have evidence of endocrine overactivity in the form of Cushing’s syndrome (pigmented nodular adrenocortical disease) and acromegaly.
The third type of localized hyperpigmentation is due to a local increase in pigment production, and it includes ephelides and café au lait macules (CALMs). While a single CALM can be seen in up to 10% of the normal population, the presence of multiple or large-sized CALMs raises the possibility of an associated genodermatosis, e.g., neurofibromatosis (NF) or McCune-Albright syndrome. CALMs are flat, uniformly brown in color (usually two shades darker than uninvolved skin), and can vary in size from 0.5–12 cm. Approximately 80–90% of adult patients with type I NF will have six or more CALMs measuring ≥1.5 cm in diameter. Additional findings are discussed in the section on neurofibromas (see “Papulonodular Skin Lesions,” below). In comparison with NF, the CALMs in patients with McCune-Albright syndrome (polyostotic fibrous dysplasia with precocious puberty in females due to mosaicism for an activating mutation in a G protein [Gsα] gene) are usually larger, are more irregular in outline, and tend to respect the midline.
In incontinentia pigmenti, dyskeratosis congenita, and bleomycin pigmentation, the areas of localized hyperpigmentation form a pattern—swirled in the first, reticulated in the second, and flagellate in the third. In dyskeratosis congenita, atrophic reticulated hyperpigmentation is seen on the neck, trunk, and thighs and is accompanied by nail dystrophy, pancytopenia, and leukoplakia of the oral and anal mucosae. The latter often develops into squamous cell carcinoma. In addition to the flagellate pigmentation (linear streaks) on the trunk, patients receiving bleomycin often have hyperpigmentation overlying the elbows, knees, and small joints of the hand.
Localized hyperpigmentation is seen as a side effect of several other systemic medications, including those that produce fixed drug reactions (nonsteroidal anti-inflammatory drugs [NSAIDs], sulfonamides, barbiturates, and tetracyclines) and those that can complex with melanin (antimalarials) or iron (minocycline). Fixed drug eruptions recur in the exact same location as circular areas of erythema that can become bullous and then resolve as brown macules. The eruption usually appears within hours of administration of the offending agent, and common locations include the genitalia, distal extremities, and perioral region. Chloroquine and hydroxychloroquine produce gray-brown to blue-black discoloration of the shins, hard palate, and face, while blue macules (often misdiagnosed as bruises) can be seen on the lower extremities and in sites of inflammation with prolonged minocycline administration. Estrogen in oral contraceptives can induce melasma—symmetric brown patches on the face, especially the cheeks, upper lip, and forehead. Similar changes are seen in pregnancy and in patients receiving phenytoin.
In the diffuse forms of hyperpigmentation, the darkening of the skin may be of equal intensity over the entire body or may be accentuated in sun-exposed areas. The causes of diffuse hyperpigmentation can be divided into four major groups—endocrine, metabolic, autoimmune, and drugs. The endocrinopathies that frequently have associated hyperpigmentation include Addison’s disease, Nelson syndrome, and ectopic ACTH syndrome. In these diseases, the increased pigmentation is diffuse but is accentuated in sun-exposed areas, the palmar creases, sites of friction, and scars. An overproduction of the pituitary hormones α-MSH (melanocyte-stimulating hormone) and ACTH can lead to an increase in melanocyte activity. These peptides are products of the proopiomelanocortin gene and exhibit homology; e.g., α-MSH and ACTH share 13 amino acids. A minority of patients with Cushing’s disease or hyperthyroidism have generalized hyperpigmentation.
The metabolic causes of hyperpigmentation include porphyria cutanea tarda (PCT), hemochromatosis, vitamin B12 deficiency, folic acid deficiency, pellagra, and malabsorption, including Whipple’s disease. In patients with PCT (see “Vesicles/Bullae,” below), the skin darkening is seen in sun-exposed areas and is a reflection of the photoreactive properties of porphyrins. The increased level of iron in the skin of patients with type 1 hemochromatosis stimulates melanin pigment production and leads to the classic bronze color. Patients with pellagra have a brown discoloration of the skin, especially in sun-exposed areas, as a result of nicotinic acid (niacin) deficiency. In the areas of increased pigmentation, there is a thin, varnish-like scale. These changes are also seen in patients who are vitamin B6 deficient, have functioning carcinoid tumors (increased consumption of niacin), or take isoniazid. Approximately 50% of the patients with Whipple’s disease have an associated generalized hyperpigmentation in association with diarrhea, weight loss, arthritis, and lymphadenopathy. A diffuse, slate-blue to gray-brown color is seen in patients with melanosis secondary to metastatic melanoma. The color reflects widespread deposition of melanin within the dermis as a result of the high concentration of circulating melanin precursors.
Of the autoimmune diseases associated with diffuse hyperpigmentation, biliary cirrhosis and systemic sclerosis are the most common, and occasionally, both disorders are seen in the same patient. The skin is dark brown in color, especially in sun-exposed areas. In biliary cirrhosis, the hyperpigmentation is accompanied by pruritus, jaundice, and xanthomas, whereas in systemic sclerosis, it is accompanied by sclerosis of the extremities, face, and, less commonly, the trunk. Additional clues to the diagnosis of systemic sclerosis are mat and periungual telangiectasias, calcinosis cutis, Raynaud’s phenomenon, and distal ulcerations (see “Telangiectasias,” above). The differential diagnosis of cutaneous sclerosis with hyperpigmentation includes the POEMS (polyneuropathy; organomegaly [liver, spleen, lymph nodes]; endocrinopathies [impotence, gynecomastia]; M-protein; and skin changes) syndrome. The skin changes include hyperpigmentation, induration, hypertrichosis, angiomas, clubbing, and facial lipoatrophy.
Diffuse hyperpigmentation that is due to drugs or metals can result from one of several mechanisms—induction of melanin pigment formation, complexing of the drug or its metabolites to melanin, and deposits of the drug in the dermis. Busulfan, cyclophosphamide, 5-fluorouracil, and inorganic arsenic induce pigment production. Complexes containing melanin or iron plus the drug or its metabolites are seen in patients receiving minocycline, and a diffuse, blue-gray, muddy appearance within sun-exposed areas may develop, in addition to pigmentation of the mucous membranes, teeth, nails, bones, and thyroid. Administration of amiodarone can result in both a phototoxic eruption (exaggerated sunburn) and/or a slate-gray to violaceous discoloration of sun-exposed skin. Biopsy specimens of the latter show yellow-brown granules in dermal macrophages, which represent intralysosomal accumulations of lipids, amiodarone, and its metabolites. Actual deposits of a particular drug or metal in the skin are seen with silver (argyria), where the skin appears blue-gray in color; gold (chrysiasis), where the skin has a brown to blue-gray color; and clofazimine, where the skin appears reddish brown. The associated pigmentation is accentuated in sun-exposed areas, and discoloration of the eye is seen with gold (sclerae) and clofazimine (conjunctivae).
VESICLES/BULLAE
(Table 72-12) Depending on their size, cutaneous blisters are referred to as vesicles (<1 cm) or bullae (>1 cm). The primary autoimmune blistering disorders include pemphigus vulgaris, pemphigus foliaceus, paraneoplastic pemphigus, bullous pemphigoid, gestational pemphigoid, cicatricial pemphigoid, epidermolysis bullosa acquisita, linear IgA bullous dermatosis (LABD), and dermatitis herpetiformis (Chap. 73).
CAUSES OF VESICLES/BULLAE |
aIntraepidermal. bSubepidermal. cAssociated with gluten enteropathy. dAssociated with inflammatory bowel disease. eDegeneration of cells within the basal layer of the epidermis can give impression split is subepidermal. fAlso systemic. gIn adults, associated with renal failure and immunocompromised state.
Vesicles and bullae are also seen in contact dermatitis, both allergic and irritant forms (Chap. 71). When there is a linear arrangement of vesicular lesions, an exogenous cause or herpes zoster should be suspected. Bullous disease secondary to the ingestion of drugs can take one of several forms, including phototoxic eruptions, isolated bullae, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (Chap. 74). Clinically, phototoxic eruptions resemble an exaggerated sunburn with diffuse erythema and bullae in sun-exposed areas. The most commonly associated drugs are doxycycline, quinolones, thiazides, NSAIDs, voriconazole, and psoralens. The development of a phototoxic eruption is dependent on the doses of both the drug and ultraviolet (UV)-A irradiation.
Toxic epidermal necrolysis is characterized by bullae that arise on widespread areas of tender erythema and then slough. This results in large areas of denuded skin. The associated morbidity, such as sepsis, and mortality rates are relatively high and are a function of the extent of epidermal necrosis. In addition, these patients may also have involvement of the mucous membranes and respiratory and intestinal tracts. Drugs are the primary cause of TEN, and the most common offenders are aromatic anticonvulsants (phenytoin, barbiturates, carbamazepine), sulfonamides, aminopenicillins, allopurinol, and NSAIDs. Severe acute graft-versus-host disease (grade 4), vancomycin-induced LABD, and the acute syndrome of apoptotic pan-epidermolysis (ASAP) in patients with lupus can also resemble TEN.
In erythema multiforme (EM), the primary lesions are pink-red macules and edematous papules, the centers of which may become vesicular. In contrast to a morbilliform exanthem, the clue to the diagnosis of EM, and especially SJS, is the development of a “dusky” violet color in the center of the lesions. Target lesions are also characteristic of EM and arise as a result of active centers and borders in combination with centrifugal spread. However, target lesions need not be present to make the diagnosis of EM.
EM has been subdivided into two major groups: (1) EM minor due to herpes simplex virus (HSV) and (2) EM major due to HSV; Mycoplasma pneumoniae; or, occasionally, drugs. Involvement of the mucous membranes (ocular, nasal, oral, and genital) is seen more commonly in the latter form. Hemorrhagic crusts of the lips are characteristic of EM major and SJS as well as herpes simplex, pemphigus vulgaris, and paraneoplastic pemphigus. Fever, malaise, myalgias, sore throat, and cough may precede or accompany the eruption. The lesions of EM usually resolve over 2–4 weeks but may be recurrent, especially when due to HSV. In addition to HSV (in which lesions usually appear 7–12 days after the viral eruption), EM can also follow vaccinations, radiation therapy, and exposure to environmental toxins, including the oleoresin in poison ivy.
Induction of SJS is most often due to drugs, especially sulfonamides, phenytoin, barbiturates, lamotrigine, aminopenicillins, nonnucleoside reverse transcriptase inhibitors (e.g., nevirapine), and carbamazepine. Widespread dusky macules and significant mucosal involvement are characteristic of SJS, and the cutaneous lesions may or may not develop epidermal detachment. If the latter occurs, by definition, it is limited to <10% of the body surface area (BSA). Greater involvement leads to the diagnosis of SJS/TEN overlap (10–30% BSA) or TEN (>30% BSA).
In addition to primary blistering disorders and hypersensitivity reactions, bacterial and viral infections can lead to vesicles and bullae. The most common infectious agents are HSV (Chap. 216), varicella-zoster virus (Chap. 217), and S. aureus (Chap. 172).
Staphylococcal scalded-skin syndrome (SSSS) and bullous impetigo are two blistering disorders associated with staphylococcal (phage group II) infection. In SSSS, the initial findings are redness and tenderness of the central face, neck, trunk, and intertriginous zones. This is followed by short-lived flaccid bullae and a slough or exfoliation of the superficial epidermis. Crusted areas then develop, characteristically around the mouth in a radial pattern. SSSS is distinguished from TEN by the following features: younger age group (primarily infants), more superficial site of blister formation, no oral lesions, shorter course, lower morbidity and mortality rates, and an association with staphylococcal exfoliative toxin (“exfoliatin”), not drugs. A rapid diagnosis of SSSS versus TEN can be made by a frozen section of the blister roof or exfoliative cytology of the blister contents. In SSSS, the site of staphylococcal infection is usually extracutaneous (conjunctivitis, rhinorrhea, otitis media, pharyngitis, tonsillitis), and the cutaneous lesions are sterile, whereas in bullous impetigo, the skin lesions are the site of infection. Impetigo is more localized than SSSS and usually presents with honey-colored crusts. Occasionally, superficial purulent blisters also form. Cutaneous emboli from gram-negative infections may present as isolated bullae, but the base of the lesion is purpuric or necrotic, and it may develop into an ulcer (see “Purpura,” below).
Several metabolic disorders are associated with blister formation, including diabetes mellitus, renal failure, and porphyria. Local hypoxemia secondary to decreased cutaneous blood flow can also produce blisters, which explains the presence of bullae over pressure points in comatose patients (coma bullae). In diabetes mellitus, tense bullae with clear sterile viscous fluid arise on normal skin. The lesions can be as large as 6 cm in diameter and are located on the distal extremities. There are several types of porphyria, but the most common form with cutaneous findings is porphyria cutanea tarda (PCT). In sun-exposed areas (primarily the face and hands), the skin is very fragile, with trauma leading to erosions mixed with tense vesicles. These lesions then heal with scarring and formation of milia; the latter are firm, 1- to 2-mm white or yellow papules that represent epidermoid inclusion cysts. Associated findings can include hypertrichosis of the lateral malar region (men) or face (women) and, in sun-exposed areas, hyperpigmentation and firm sclerotic plaques. An elevated level of urinary uroporphyrins confirms the diagnosis and is due to a decrease in uroporphyrinogen decarboxylase activity. PCT can be exacerbated by alcohol, hemochromatosis and other forms of iron overload, chlorinated hydrocarbons, hepatitis C and HIV infections, and hepatomas.
The differential diagnosis of PCT includes (1) porphyria variegata—the skin signs of PCT plus the systemic findings of acute intermittent porphyria; it has a diagnostic plasma porphyrin fluorescence emission at 626 nm; (2) drug-induced pseudoporphyria—the clinical and histologic findings are similar to PCT, but porphyrins are normal; etiologic agents include naproxen and other NSAIDs, furosemide, tetracycline, and voriconazole; (3) bullous dermatosis of hemodialysis—the same appearance as PCT, but porphyrins are usually normal or occasionally borderline elevated; patients have chronic renal failure and are on hemodialysis; (4) PCT associated with hepatomas and hemodialysis; and (5) epidermolysis bullosa acquisita (Chap. 73).
EXANTHEMS
(Table 72-13) Exanthems are characterized by an acute generalized eruption. The most common presentation is erythematous macules and papules (morbilliform) and less often confluent blanching erythema (scarlatiniform). Morbilliform eruptions are usually due to either drugs or viral infections. For example, up to 5% of patients receiving penicillins, sulfonamides, phenytoin, or nevirapine will develop a maculopapular eruption. Accompanying signs may include pruritus, fever, eosinophilia, and transient lymphadenopathy. Similar maculopapular eruptions are seen in the classic childhood viral exanthems, including (1) rubeola (measles)—a prodrome of coryza, cough, and conjunctivitis followed by Koplik’s spots on the buccal mucosa; the eruption begins behind the ears, at the hairline, and on the forehead and then spreads down the body, often becoming confluent; (2) rubella—the eruption begins on the forehead and face and then spreads down the body; it resolves in the same order and is associated with retroauricular and suboccipital lymphadenopathy; and (3) erythema infectiosum (fifth disease)—erythema of the cheeks is followed by a reticulated pattern on the extremities; it is secondary to a parvovirus B19 infection, and an associated arthritis is seen in adults.
CAUSES OF EXANTHEMS |
aPrimary infection in infants and reactivation in the setting of immunosuppression.
Abbreviations: CMV, cytomegalovirus; HHV, human herpesvirus; HIV, human immunodeficiency virus.
Both measles and rubella can occur in unvaccinated adults, and an atypical form of measles is seen in adults immunized with either killed measles vaccine or killed vaccine followed in time by live vaccine. In contrast to classic measles, the eruption of atypical measles begins on the palms, soles, wrists, and ankles, and the lesions may become purpuric. The patient with atypical measles can have pulmonary involvement and be quite ill. Rubelliform and roseoliform eruptions are also associated with Epstein-Barr virus (5–15% of patients), echovirus, coxsackievirus, cytomegalovirus, adenovirus, dengue virus, and West Nile virus infections. Detection of specific IgM antibodies or fourfold elevations in IgG antibodies allow the proper diagnosis, but polymerase chain reaction (PCR) is gradually replacing serologic assays. Occasionally, a maculopapular drug eruption is a reflection of an underlying viral infection. For example, ~95% of the patients with infectious mononucleosis who are given ampicillin will develop a rash.
Of note, early in the course of infections with Rickettsia and meningococcus, prior to the development of petechiae and purpura, the lesions may be erythematous macules and papules. This is also the case in chickenpox prior to the development of vesicles. Maculopapular eruptions are associated with early HIV infection, early secondary syphilis, typhoid fever, and acute graft-versus-host disease. In the last, lesions frequently begin on the dorsal hands and forearms; the macular rose spots of typhoid fever involve primarily the anterior trunk.
The prototypic scarlatiniform eruption is seen in scarlet fever and is due to an erythrogenic toxin produced by bacteriophage-containing group A β-hemolytic streptococci, most commonly in the setting of pharyngitis. This eruption is characterized by diffuse erythema, which begins on the neck and upper trunk, and red follicular puncta. Additional findings include a white strawberry tongue (white coating with red papillae) followed by a red strawberry tongue (red tongue with red papillae); petechiae of the palate; a facial flush with circumoral pallor; linear petechiae in the antecubital fossae; and desquamation of the involved skin, palms, and soles 5–20 days after onset of the eruption. A similar desquamation of the palms and soles is seen in toxic shock syndrome (TSS), in Kawasaki disease, and after severe febrile illnesses. Certain strains of staphylococci also produce an erythrotoxin that leads to the same clinical findings as in streptococcal scarlet fever, except that the anti-streptolysin O or -DNase B titers are not elevated.
In toxic shock syndrome, staphylococcal (phage group I) infections produce an exotoxin (TSST-1) that causes the fever and rash as well as enterotoxins. Initially, the majority of cases were reported in menstruating women who were using tampons. However, other sites of infection, including wounds and nasal packing, can lead to TSS. The diagnosis of TSS is based on clinical criteria (Chap. 172), and three of these involve mucocutaneous sites (diffuse erythema of the skin, desquamation of the palms and soles 1–2 weeks after onset of illness, and involvement of the mucous membranes). The latter is characterized as hyperemia of the vagina, oropharynx, or conjunctivae. Similar systemic findings have been described in streptococcal toxic shock syndrome (Chap. 173), and although an exanthem is seen less often than in TSS due to a staphylococcal infection, the underlying infection is often in the soft tissue (e.g., cellulitis).
The cutaneous eruption in Kawasaki disease (Chap. 385) is polymorphous, but the two most common forms are morbilliform and scarlatiniform. Additional mucocutaneous findings include bilateral conjunctival injection; erythema and edema of the hands and feet followed by desquamation; and diffuse erythema of the oropharynx, red strawberry tongue, and dry fissured lips. This clinical picture can resemble TSS and scarlet fever, but clues to the diagnosis of Kawasaki disease are cervical lymphadenopathy, cheilitis, and thrombocytosis. The most serious associated systemic finding in this disease is coronary aneurysms secondary to arteritis. Scarlatiniform eruptions are also seen in the early phase of SSSS (see “Vesicles/Bullae,” above), in young adults with Arcanobacterium haemolyticum infection, and as reactions to drugs.
URTICARIA
(Table 72-14) Urticaria (hives) are transient lesions that are composed of a central wheal surrounded by an erythematous halo or flare. Individual lesions are round, oval, or figurate and are often pruritic. Acute and chronic urticaria have a wide variety of allergic etiologies and reflect edema in the dermis. Urticarial lesions can also be seen in patients with mastocytosis (urticaria pigmentosa), hypo- or hyperthyroidism, and systemic-onset juvenile idiopathic arthritis (Still’s disease). In both juvenile- and adult-onset Still’s disease, the lesions coincide with the fever spike, are transient, and are due to dermal infiltrates of neutrophils.
CAUSES OF URTICARIA AND ANGIOEDEMA |
aA small minority develop anaphylaxis. bAlso systemic. cAcquired angioedema can be idiopathic, associated with a lymphoproliferative disorder, or due to a drug, e.g., angiotensin-converting enzyme (ACE) inhibitors.
The common physical urticarias include dermatographism, solar urticaria, cold urticaria, and cholinergic urticaria. Patients with dermatographism exhibit linear wheals following minor pressure or scratching of the skin. It is a common disorder, affecting ~5% of the population. Solar urticaria characteristically occurs within minutes of sun exposure and is a skin sign of one systemic disease—erythropoietic protoporphyria. In addition to the urticaria, these patients have subtle pitted scarring of the nose and hands. Cold urticaria is precipitated by exposure to the cold, and therefore exposed areas are usually affected. In occasional patients, the disease is associated with abnormal circulating proteins—more commonly cryoglobulins and less commonly cryofibrinogens. Additional systemic symptoms include wheezing and syncope, thus explaining the need for these patients to avoid swimming in cold water. Autosomal dominantly inherited cold urticaria is associated with dysfunction of cryopyrin. Cholinergic urticaria is precipitated by heat, exercise, or emotion and is characterized by small wheals with relatively large flares. It is occasionally associated with wheezing.
Whereas urticarias are the result of dermal edema, subcutaneous edema leads to the clinical picture of angioedema. Sites of involvement include the eyelids, lips, tongue, larynx, and gastrointestinal tract as well as the subcutaneous tissue. Angioedema occurs alone or in combination with urticaria, including urticarial vasculitis and the physical urticarias. Both acquired and hereditary (autosomal dominant) forms of angioedema occur (Chap. 376), and in the latter, urticaria is rarely, if ever, seen.
Urticarial vasculitis is an immune complex disease that may be confused with simple urticaria. In contrast to simple urticaria, individual lesions tend to last longer than 24 h and usually develop central petechiae that can be observed even after the urticarial phase has resolved. The patient may also complain of burning rather than pruritus. On biopsy, there is a leukocytoclastic vasculitis of the small dermal blood vessels. Although many cases of urticarial vasculitis are idiopathic in origin, it can be a reflection of an underlying systemic illness such as lupus erythematosus, Sjögren’s syndrome, or hereditary complement deficiency. There is a spectrum of urticarial vasculitis that ranges from purely cutaneous to multisystem involvement. The most common systemic signs and symptoms are arthralgias and/or arthritis, nephritis, and crampy abdominal pain, with asthma and chronic obstructive lung disease seen less often. Hypocomplementemia occurs in one- to two-thirds of patients, even in the idiopathic cases. Urticarial vasculitis can also be seen in patients with hepatitis B and hepatitis C infections, serum sickness, and serum sickness–like illnesses (e.g., due to cefaclor, minocycline).
PAPULONODULAR SKIN LESIONS
(Table 72-15) In the papulonodular diseases, the lesions are elevated above the surface of the skin and may coalesce to form larger plaques. The location, consistency, and color of the lesions are the keys to their diagnosis; this section is organized on the basis of color.
PAPULONODULAR SKIN LESIONS ACCORDING TO COLOR GROUPS |
aIf multiple with childhood onset, consider Gardner syndrome. bMay have darker hue in more darkly pigmented individuals. cSee also “Hyperpigmentation.”
Abbreviation: MEN, multiple endocrine neoplasia.
WHITE LESIONS
In calcinosis cutis, there are firm white to white-yellow papules with an irregular surface. When the contents are expressed, a chalky white material is seen. Dystrophic calcification is seen at sites of previous inflammation or damage to the skin. It develops in acne scars as well as on the distal extremities of patients with systemic sclerosis and in the subcutaneous tissue and intermuscular fascial planes in DM. The latter is more extensive and is more commonly seen in children. An elevated calcium phosphate product, most commonly due to secondary hyperparathyroidism in the setting of renal failure, can lead to nodules of metastatic calcinosis cutis, which tend to be subcutaneous and periarticular. These patients can also develop calcification of muscular arteries and subsequent ischemic necrosis (calciphylaxis). Osteoma cutis, in the form of small papules, most commonly occurs on the face of individuals with a history of acne vulgaris, whereas plate-like lesions occur in rare genetic syndromes (Chap. 82).
SKIN-COLORED LESIONS
There are several types of skin-colored lesions, including epidermoid inclusion cysts, lipomas, rheumatoid nodules, neurofibromas, angiofibromas, neuromas, and adnexal tumors such as tricholemmomas. Both epidermoid inclusion cysts and lipomas are very common mobile subcutaneous nodules—the former are rubbery and drain cheeselike material (sebum and keratin) if incised. Lipomas are firm and somewhat lobulated on palpation. When extensive facial epidermoid inclusion cysts develop during childhood or there is a family history of such lesions, the patient should be examined for other signs of Gardner syndrome, including osteomas and desmoid tumors. Rheumatoid nodules are firm 0.5- to 4-cm nodules that favor the extensor aspect of joints, especially the elbows. They are seen in ~20% of patients with rheumatoid arthritis and 6% of patients with Still’s disease. Biopsies of the nodules show palisading granulomas. Similar lesions that are smaller and shorter-lived are seen in rheumatic fever.
Neurofibromas (benign Schwann cell tumors) are soft papules or nodules that exhibit the “button-hole” sign; that is, they invaginate into the skin with pressure in a manner similar to a hernia. Single lesions are seen in normal individuals, but multiple neurofibromas, usually in combination with six or more CALMs measuring >1.5 cm (see “Hyperpigmentation,” above), axillary freckling, and multiple Lisch nodules, are seen in von Recklinghausen’s disease (NF type I) (Chap. 118). In some patients, the neurofibromas are localized and unilateral due to somatic mosaicism.
Angiofibromas are firm pink to skin-colored papules that measure from 3 mm to a few centimeters in diameter. When multiple lesions are located on the central cheeks (adenoma sebaceum), the patient has tuberous sclerosis or multiple endocrine neoplasia (MEN) syndrome, type 1. The former is an autosomal disorder due to mutations in two different genes, and the associated findings are discussed in the section on ash leaf spots as well as in Chap. 118.
Neuromas (benign proliferations of nerve fibers) are also firm, skin-colored papules. They are more commonly found at sites of amputation and as rudimentary supernumerary digits. However, when there are multiple neuromas on the eyelids, lips, distal tongue, and/or oral mucosa, the patient should be investigated for other signs of the MEN syndrome, type 2b. Associated findings include marfanoid habitus, protuberant lips, intestinal ganglioneuromas, and medullary thyroid carcinoma (>75% of patients; Chap. 408).
Adnexal tumors are derived from pluripotent cells of the epidermis that can differentiate toward hair, sebaceous, apocrine, or eccrine glands or remain undifferentiated. Basal cell carcinomas (BCCs) are examples of adnexal tumors that have little or no evidence of differentiation. Clinically, they are translucent papules with rolled borders, telangiectasias, and central erosion. BCCs commonly arise in sun-damaged skin of the head and neck as well as the upper trunk. When a patient has multiple BCCs, especially prior to age 30, the possibility of the nevoid basal cell carcinoma syndrome should be raised. It is inherited as an autosomal dominant trait and is associated with jaw cysts, palmar and plantar pits, frontal bossing, medulloblastomas, and calcification of the falx cerebri and diaphragma sellae. Tricholemmomas are also skin-colored adnexal tumors but differentiate toward hair follicles and can have a wartlike appearance. The presence of multiple tricholemmomas on the face and cobblestoning of the oral mucosa points to the diagnosis of Cowden disease (multiple hamartoma syndrome) due to mutations in the phosphatase and tensin homolog (PTEN) gene. Internal organ involvement (in decreasing order of frequency) includes fibrocystic disease and carcinoma of the breast, adenomas and carcinomas of the thyroid, and gastrointestinal polyposis. Keratoses of the palms, soles, and dorsal aspect of the hands are also seen.
PINK LESIONS
The cutaneous lesions associated with primary systemic amyloidosis are often pink in color and translucent. Common locations are the face, especially the periorbital and perioral regions, and flexural areas. On biopsy, homogeneous deposits of amyloid are seen in the dermis and in the walls of blood vessels; the latter lead to an increase in vessel wall fragility. As a result, petechiae and purpura develop in clinically normal skin as well as in lesional skin following minor trauma, hence the term pinch purpura. Amyloid deposits are also seen in the striated muscle of the tongue and result in macroglossia.
Even though specific mucocutaneous lesions are present in only ~30% of the patients with primary systemic (AL) amyloidosis, the diagnosis can be made via histologic examination of abdominal subcutaneous fat, in conjunction with a serum free light chain assay. By special staining, amyloid deposits are seen around blood vessels or individual fat cells in 40–50% of patients. There are also three forms of amyloidosis that are limited to the skin and that should not be construed as cutaneous lesions of systemic amyloidosis. They are macular amyloidosis (upper back), lichen amyloidosis (usually lower extremities), and nodular amyloidosis. In macular and lichen amyloidosis, the deposits are composed of altered epidermal keratin. Early-onset macular and lichen amyloidosis have been associated with MEN syndrome, type 2a.
Patients with multicentric reticulohistiocytosis also have pink-colored papules and nodules on the face and mucous membranes as well as on the extensor surface of the hands and forearms. They have a polyarthritis that can mimic rheumatoid arthritis clinically. On histologic examination, the papules have characteristic giant cells that are not seen in biopsies of rheumatoid nodules. Pink to skin-colored papules that are firm, 2–5 mm in diameter, and often in a linear arrangement are seen in patients with papular mucinosis. This disease is also referred to as generalized lichen myxedematosus or scleromyxedema. The latter name comes from the induration of the face and extremities that may accompany the papular eruption. Biopsy specimens of the papules show localized mucin deposition, and serum protein electrophoresis plus immunofixation electrophoresis demonstrates a monoclonal spike of IgG, usually with a λ light chain.
YELLOW LESIONS
Several systemic disorders are characterized by yellow-colored cutaneous papules or plaques—hyperlipidemia (xanthomas), gout (tophi), diabetes (necrobiosis lipoidica), pseudoxanthoma elasticum, and Muir-Torre syndrome (sebaceous tumors). Eruptive xanthomas are the most common form of xanthomas and are associated with hypertriglyceridemia (primarily hyperlipoproteinemia types I, IV, and V). Crops of yellow papules with erythematous halos occur primarily on the extensor surfaces of the extremities and the buttocks, and they spontaneously involute with a fall in serum triglycerides. Types II and III result in one or more of the following types of xanthoma: xanthelasma, tendon xanthomas, and plane xanthomas. Xanthelasma are found on the eyelids, whereas tendon xanthomas are frequently associated with the Achilles and extensor finger tendons; plane xanthomas are flat and favor the palmar creases, neck, upper trunk, and flexural folds. Tuberous xanthomas are frequently associated with hypertriglyceridemia, but they are also seen in patients with hypercholesterolemia and are found most frequently over the large joints or hand. Biopsy specimens of xanthomas show collections of lipid-containing macrophages (foam cells).
Patients with several disorders, including biliary cirrhosis, can have a secondary form of hyperlipidemia with associated tuberous and plane xanthomas. However, patients with plasma cell dyscrasias have normolipemic plane xanthomas. This latter form of xanthoma may be ≥12 cm in diameter and is most frequently seen on the upper trunk or side of the neck. It is important to note that the most common setting for eruptive xanthomas is uncontrolled diabetes mellitus. The least specific sign for hyperlipidemia is xanthelasma, because at least 50% of the patients with this finding have normal lipid profiles.
In tophaceous gout, there are deposits of monosodium urate in the skin around the joints, particularly those of the hands and feet. Additional sites of tophi formation include the helix of the ear and the olecranon and prepatellar bursae. The lesions are firm, yellow in color, and occasionally discharge a chalky material. Their size varies from 1 mm to 7 cm, and the diagnosis can be established by polarized light microscopy of the aspirated contents of a lesion. Lesions of necrobiosis lipoidica are found primarily on the shins (90%), and patients can have diabetes mellitus or develop it subsequently. Characteristic findings include a central yellow color, atrophy (transparency), telangiectasias, and a red to red-brown border. Ulcerations can also develop within the plaques. Biopsy specimens show necrobiosis of collagen and granulomatous inflammation.
In pseudoxanthoma elasticum (PXE), due to mutations in the gene ABCC6, there is an abnormal deposition of calcium on the elastic fibers of the skin, eye, and blood vessels. In the skin, the flexural areas such as the neck, axillae, antecubital fossae, and inguinal area are the primary sites of involvement. Yellow papules coalesce to form reticulated plaques that have an appearance similar to that of plucked chicken skin. In severely affected skin, hanging, redundant folds develop. Biopsy specimens of involved skin show swollen and irregularly clumped elastic fibers with deposits of calcium. In the eye, the calcium deposits in Bruch’s membrane lead to angioid streaks and choroiditis; in the arteries of the heart, kidney, gastrointestinal tract, and extremities, the deposits lead to angina, hypertension, gastrointestinal bleeding, and claudication, respectively.
Adnexal tumors that have differentiated toward sebaceous glands include sebaceous adenoma, sebaceous carcinoma, and sebaceous hyperplasia. Except for sebaceous hyperplasia, which is commonly seen on the face, these tumors are fairly rare. Patients with Muir-Torre syndrome have one or more sebaceous adenoma(s), and they can also have sebaceous carcinomas and sebaceous hyperplasia as well as keratoacanthomas. The internal manifestations of Muir-Torre syndrome include multiple carcinomas of the gastrointestinal tract (primarily colon) as well as cancers of the larynx, genitourinary tract, and breast.
RED LESIONS
Cutaneous lesions that are red in color have a wide variety of etiologies; in an attempt to simplify their identification, they will be subdivided into papules, papules/plaques, and subcutaneous nodules. Common red papules include arthropod bites and cherry hemangiomas; the latter are small, bright-red, dome-shaped papules that represent a benign proliferation of capillaries. In patients with AIDS (Chap. 226), the development of multiple red hemangioma-like lesions points to bacillary angiomatosis, and biopsy specimens show clusters of bacilli that stain positive with the Warthin-Starry stain; the pathogens have been identified as Bartonella henselae and Bartonella quintana. Disseminated visceral disease is seen primarily in immunocompromised hosts but can occur in immunocompetent individuals.
Multiple angiokeratomas are seen in Fabry disease, an X-linked recessive lysosomal storage disease that is due to a deficiency of α-galactosidase A. The lesions are red to red-blue in color and can be quite small in size (1–3 mm), with the most common location being the lower trunk. Associated findings include chronic renal disease, peripheral neuropathy, and corneal opacities (cornea verticillata). Electron photomicrographs of angiokeratomas and clinically normal skin demonstrate lamellar lipid deposits in fibroblasts, pericytes, and endothelial cells that are diagnostic of this disease. Widespread acute eruptions of erythematous papules are discussed in the section on exanthems.
There are several infectious diseases that present as erythematous papules or nodules in a lymphocutaneous or sporotrichoid pattern, i.e., in a linear arrangement along the lymphatic channels. The two most common etiologies are Sporothrix schenckii (sporotrichosis) and the atypical mycobacterium Mycobacterium marinum. The organisms are introduced as a result of trauma, and a primary inoculation site is often seen in addition to the lymphatic nodules. Additional causes include Nocardia, Leishmania, and other atypical mycobacteria and dimorphic fungi; culture of lesional tissue will aid in the diagnosis.
The diseases that are characterized by erythematous plaques with scale are reviewed in the papulosquamous section, and the various forms of dermatitis are discussed in the section on erythroderma. Additional disorders in the differential diagnosis of red papules/plaques include cellulitis, polymorphous light eruption (PMLE), cutaneous lymphoid hyperplasia (lymphocytoma cutis), cutaneous lupus, lymphoma cutis, and leukemia cutis. The first three diseases represent primary cutaneous disorders, although cellulitis may be accompanied by a bacteremia. PMLE is characterized by erythematous papules and plaques in a primarily sun-exposed distribution—dorsum of the hand, extensor forearm, and upper trunk. Lesions follow exposure to UV-B and/or UV-A, and in higher latitudes, PMLE is most severe in the late spring and early summer. A process referred to as “hardening” occurs with continued UV exposure, and the eruption fades, but in temperate climates, it will recur in the spring. PMLE must be differentiated from cutaneous lupus, and this is accomplished by observation of the natural history, histologic examination, and direct immunofluorescence of the lesions. Cutaneous lymphoid hyperplasia (pseudolymphoma) is a benign polyclonal proliferation of lymphocytes in the skin that presents as infiltrated pink-red to red-purple papules and plaques; it must be distinguished from lymphoma cutis.
Several types of red plaques are seen in patients with systemic lupus, including (1) erythematous urticarial plaques across the cheeks and nose in the classic butterfly rash; (2) erythematous discoid lesions with fine or “carpet-tack” scale, telangiectasias, central hypopigmentation, peripheral hyperpigmentation, follicular plugging, and atrophy located on the face, scalp, external ears, arms, and upper trunk; and (3) psoriasiform or annular lesions of subacute cutaneous lupus with hypopigmented centers located primarily on the extensor arms and upper trunk. Additional mucocutaneous findings include (1) a violaceous flush on the face and V of the neck; (2) photosensitivity; (3) urticarial vasculitis (see “Urticaria,” above); (4) lupus panniculitis (see below); (5) diffuse alopecia; (6) alopecia secondary to discoid lesions; (7) periungual telangiectasias and erythema; (8) EM-like lesions that may become bullous; (9) oral ulcers; and (10) distal ulcerations secondary to Raynaud’s phenomenon, vasculitis, or livedoid vasculopathy. Patients with only discoid lesions usually have the form of lupus that is limited to the skin. However, up to 10% of these patients eventually develop systemic lupus. Direct immunofluorescence of involved skin, in particular discoid lesions, shows deposits of IgG or IgM and C3 in a granular distribution along the dermal-epidermal junction.
In lymphoma cutis, there is a proliferation of malignant lymphocytes in the skin, and the clinical appearance resembles that of cutaneous lymphoid hyperplasia—infiltrated pink-red to red-purple papules and plaques. Lymphoma cutis can occur anywhere on the surface of the skin, whereas the sites of predilection for lymphocytomas include the malar ridge, tip of the nose, and earlobes. Patients with non-Hodgkin’s lymphomas have specific cutaneous lesions more often than those with Hodgkin’s disease, and, occasionally, the skin nodules precede the development of extracutaneous non-Hodgkin’s lymphoma or represent the only site of involvement (e.g., primary cutaneous B cell lymphoma). Arcuate lesions are sometimes seen in lymphoma and lymphocytoma cutis as well as in CTCL. Adult T cell leukemia/lymphoma that develops in association with HTLV-1 infection is characterized by cutaneous plaques, hypercalcemia, and circulating CD25+ lymphocytes. Leukemia cutis has the same appearance as lymphoma cutis, and specific lesions are seen more commonly in monocytic leukemias than in lymphocytic or granulocytic leukemias. Cutaneous chloromas (granulocytic sarcomas) may precede the appearance of circulating blasts in acute myelogenous leukemia and, as such, represent a form of aleukemic leukemia cutis.
Sweet syndrome is characterized by pink-red to red-brown edematous plaques that are frequently painful and occur primarily on the head, neck, and upper (and, less often, lower) extremities. The patients also have fever, neutrophilia, and a dense dermal infiltrate of neutrophils in the lesions. In ~10% of the patients, there is an associated malignancy, most commonly acute myelogenous leukemia. Sweet syndrome has also been reported with inflammatory bowel disease, systemic lupus erythematosus, and solid tumors (primarily of the genitourinary tract) as well as drugs (e.g., all-trans-retinoic acid, granulocyte colony-stimulating factor [G-CSF]). The differential diagnosis includes neutrophilic eccrine hidradenitis; bullous forms of pyoderma gangrenosum; and, occasionally, cellulitis. Extracutaneous sites of involvement include joints, muscles, eye, kidney (proteinuria, occasionally glomerulonephritis), and lung (neutrophilic infiltrates). The idiopathic form of Sweet syndrome is seen more often in women, following a respiratory tract infection.
Common causes of erythematous subcutaneous nodules include inflamed epidermoid inclusion cysts, acne cysts, and furuncles. Panniculitis, an inflammation of the fat, also presents as subcutaneous nodules and is frequently a sign of systemic disease. There are several forms of panniculitis, including erythema nodosum, erythema induratum/nodular vasculitis, lupus panniculitis, lipodermatosclerosis, α1-antitrypsin deficiency, factitial, and fat necrosis secondary to pancreatic disease. Except for erythema nodosum, these lesions may break down and ulcerate or heal with a scar. The shin is the most common location for the nodules of erythema nodosum, whereas the calf is the most common location for lesions of erythema induratum. In erythema nodosum, the nodules are initially red but then develop a blue color as they resolve. Patients with erythema nodosum but no underlying systemic illness can still have fever, malaise, leukocytosis, arthralgias, and/or arthritis. However, the possibility of an underlying illness should be excluded, and the most common associations are streptococcal infections, upper respiratory viral infections, sarcoidosis, and inflammatory bowel disease, in addition to drugs (oral contraceptives, sulfonamides, penicillins, bromides, iodides). Less common associations include bacterial gastroenteritis (Yersinia, Salmonella) and coccidioidomycosis followed by tuberculosis, histoplasmosis, brucellosis, and infections with Chlamydophila pneumoniae or Chlamydia trachomatis, Mycoplasma pneumoniae, or hepatitis B virus.
Erythema induratum and nodular vasculitis have overlapping features clinically and histologically, and whether they represent two separate entities or the ends of a single disease spectrum is a point of debate; in general, the latter is usually idiopathic and the former is associated with the presence of Mycobacterium tuberculosis DNA by PCR within skin lesions. The lesions of lupus panniculitis are found primarily on the cheeks, upper arms, and buttocks (sites of abundant fat) and are seen in both the cutaneous and systemic forms of lupus. The overlying skin may be normal, erythematous, or have the changes of discoid lupus. The subcutaneous fat necrosis that is associated with pancreatic disease is presumably secondary to circulating lipases and is seen in patients with pancreatic carcinoma as well as in patients with acute and chronic pancreatitis. In this disorder, there may be an associated arthritis, fever, and inflammation of visceral fat. Histologic examination of deep incisional biopsy specimens will aid in the diagnosis of the particular type of panniculitis.
Subcutaneous erythematous nodules are also seen in cutaneous polyarteritis nodosa and as a manifestation of systemic vasculitis when there is involvement of medium-sized vessels, e.g., systemic polyarteritis nodosa, allergic granulomatosis, or granulomatosis with polyangiitis (Wegener’s) (Chap. 385). Cutaneous polyarteritis nodosa presents with painful subcutaneous nodules and ulcers within a red-purple, netlike pattern of livedo reticularis. The latter is due to slowed blood flow through the superficial horizontal venous plexus. The majority of lesions are found on the lower extremities, and while arthralgias and myalgias may accompany cutaneous polyarteritis nodosa, there is no evidence of systemic involvement. In both the cutaneous and systemic forms of vasculitis, skin biopsy specimens of the associated nodules will show the changes characteristic of a necrotizing vasculitis and/or granulomatous inflammation.
RED-BROWN LESIONS
The cutaneous lesions in sarcoidosis (Chap. 390) are classically red to red-brown in color, and with diascopy (pressure with a glass slide), a yellow-brown residual color is observed that is secondary to the granulomatous infiltrate. The waxy papules and plaques may be found anywhere on the skin, but the face is the most common location. Usually there are no surface changes, but occasionally the lesions will have scale. Biopsy specimens of the papules show “naked” granulomas in the dermis, i.e., granulomas surrounded by a minimal number of lymphocytes. Other cutaneous findings in sarcoidosis include annular lesions with an atrophic or scaly center, papules within scars, hypopigmented papules and patches, alopecia, acquired ichthyosis, erythema nodosum, and lupus pernio (see below).
The differential diagnosis of sarcoidosis includes foreign-body granulomas produced by chemicals such as beryllium and zirconium, late secondary syphilis, and lupus vulgaris. Lupus vulgaris is a form of cutaneous tuberculosis that is seen in previously infected and sensitized individuals. There is often underlying active tuberculosis elsewhere, usually in the lungs or lymph nodes. Lesions occur primarily in the head and neck region and are red-brown plaques with a yellow-brown color on diascopy. Secondary scarring and squamous cell carcinomas can develop within the plaques. Cultures or PCR analysis of the lesions should be performed, along with an interferon γ release assay of peripheral blood, because it is rare for the acid-fast stain to show bacilli within the dermal granulomas.
A generalized distribution of red-brown macules and papules is seen in the form of mastocytosis known as urticaria pigmentosa (Chap. 376). Each lesion represents a collection of mast cells in the dermis, with hyperpigmentation of the overlying epidermis. Stimuli such as rubbing cause these mast cells to degranulate, and this leads to the formation of localized urticaria (Darier’s sign). Additional symptoms can result from mast cell degranulation and include headache, flushing, diarrhea, and pruritus. Mast cells also infiltrate various organs such as the liver, spleen, and gastrointestinal tract, and accumulations of mast cells in the bones may produce either osteosclerotic or osteolytic lesions on radiographs. In the majority of these patients, however, the internal involvement remains indolent. A subtype of chronic cutaneous small-vessel vasculitis, erythema elevatum diutinum (EED), also presents with papules that are red-brown in color. The papules coalesce into plaques on the extensor surfaces of knees, elbows, and the small joints of the hand. Flares of EED have been associated with streptococcal infections.
BLUE LESIONS
Lesions that are blue in color are the result of vascular ectasias, hyperplasias and tumors or melanin pigment within the dermis. Venous lakes (ectasias) are compressible dark-blue lesions that are found commonly in the head and neck region. Venous malformations are also compressible blue papulonodules and plaques that can occur anywhere on the body, including the oral mucosa. When there are multiple rather than single congenital lesions, the patient may have the blue rubber bleb syndrome or Maffucci’s syndrome. Patients with the blue rubber bleb syndrome also have vascular anomalies of the gastrointestinal tract that may bleed, whereas patients with Maffucci’s syndrome have associated osteochondromas. Blue nevi (moles) are seen when there are collections of pigment-producing nevus cells in the dermis. These benign papular lesions are dome-shaped and occur most commonly on the dorsum of the hand or foot or in the head and neck region.
VIOLACEOUS LESIONS
Violaceous papules and plaques are seen in lupus pernio, lymphoma cutis, and cutaneous lupus. Lupus pernio is a particular type of sarcoidosis that involves the tip and alar rim of the nose as well as the earlobes, with lesions that are violaceous in color rather than red-brown. This form of sarcoidosis is associated with involvement of the upper respiratory tract. The plaques of lymphoma cutis and cutaneous lupus may be red or violaceous in color and were discussed above.
PURPLE LESIONS
Purple-colored papules and plaques are seen in vascular tumors, such as Kaposi’s sarcoma (Chap. 226) and angiosarcoma, and when there is extravasation of red blood cells into the skin in association with inflammation, as in palpable purpura (see “Purpura,” below). Patients with congenital or acquired AV fistulas and venous hypertension can develop purple papules on the lower extremities that can resemble Kaposi’s sarcoma clinically and histologically; this condition is referred to as pseudo-Kaposi’s sarcoma (acral angiodermatitis). Angiosarcoma is found most commonly on the scalp and face of elderly patients or within areas of chronic lymphedema and presents as purple papules and plaques. In the head and neck region, the tumor often extends beyond the clinically defined borders and may be accompanied by facial edema.
BROWN AND BLACK LESIONS
Brown- and black-colored papules are reviewed in “Hyperpigmentation,” above.
CUTANEOUS METASTASES
These are discussed last because they can have a wide range of colors. Most commonly, they present as either firm, skin-colored subcutaneous nodules or firm, red to red-brown papulonodules. The lesions of lymphoma cutis range from pink-red to plum in color, whereas metastatic melanoma can be pink, blue, or black in color. Cutaneous metastases develop from hematogenous or lymphatic spread and are most often due to the following primary carcinomas: in men, melanoma, oropharynx, lung, and colon; and in women, breast, melanoma, and ovary. These metastatic lesions may be the initial presentation of the carcinoma, especially when the primary site is the lung.
PURPURA
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


