Chapter 14 Cutaneous adverse reactions to drugs
Adverse drug reactions – introduction
Adverse drug reactions are unintended and undesired effects of drugs used for prevention, diagnosis or treatment of disease.1,2 In light of the ever-increasing number of medications available, it should come as no surprise that such reactions are extremely common. The incidence statistics vary considerably depending upon the method by which the data are derived and the nature of the population under study.3 Estimates, however, range from 2% to 7% of hospital inpatients.4–8 Although most reactions are mild, they are sometimes severe and a source of considerable morbidity and occasional mortality.6,7
The diagnosis of an adverse drug reaction is frequently problematical, the clinical appearances often being similar, if not identical, to a number of primary dermatoses and infectious conditions (particularly viral exanthems) and, in the context of transplantation patients, graft-versus-host disease (GVHD). The histological diagnosis can also be extremely difficult, as drug reactions can demonstrate several inflammatory histological patterns that mimic other dermatoses (i.e. spongiotic, psoriasiform, lichenoid, pityriasiform).9 The problem is exacerbated in the immunologically compromised patient. Frequently, the diagnostic difficulties are worsened by the multitude of drugs prescribed. The problem is further compounded by the multiplicity of different eruptions that any one particular drug may induce. Contrariwise, a given clinical appearance may be caused by a large number of unrelated drugs.10
The prevalence of agents responsible for adverse drug reactions reflects the prescribing tendencies for any given population as much as the relative risks ascribed to any particular drug.11 It should come as no surprise, therefore, that – in a hospital environment – antibiotics, nonsteroidal antiinflammatory agents (NSAIDs) and psychotropic drugs are commonly reported as being the most frequently incriminated.8 In a large hospital survey, penicillin and sulfonamides accounted for over 80% of all adverse drug reactions.8 Experience in general practice has been much less often documented. In a survey from the Netherlands, sulfonamide-trimethoprim combinations, fluoroquinolones, and penicillin were the most common antibacterials causing drug-related eruptions.3 In the series of approximately 150 000 patients, 1% developed a reaction.
Adverse drug reactions are mostly nonimmunologically mediated. They develop either as a result of an unwanted but known property of the drug (and hence are entirely predictable) or as a consequence of drug intolerance/idiosyncrasy (and are completely unpredictable).5,12–15 The former are by far the more common, accounting for approximately 80% of all adverse drug reactions. Less often, adverse drug reactions represent a manifestation of an immunological phenomenon, so-called allergic drug reactions.5,15 Although in theory the above subdivisions are sharply defined, in many patients the underlying pathogenetic mechanisms are far from clear.13
Adverse drug reactions are particularly encountered in certain population groups, for example the elderly, females, patients with Sjögren’s syndrome, and those suffering from the effects of immune deficiency including patients receiving immunosuppressive therapy and those suffering from the acquired immunodeficiency syndrome (AIDS).5
Adverse drug reactions can be divided into three categories: type A, type B, and type C.1,12,16
Type A drug reactions
Type A reactions, which are predictable and are related to the pharmacological actions or metabolism of the drug, include:1
Side effects
Side effects, which occur with almost all drugs, represent unwanted pharmacological actions. For example, methotrexate, cyclophosphamide and nitrosourea commonly result in anagen alopecia by inducing Bax protein-mediated apoptosis.17–21 Gold may be associated with cutaneous pigmentation (chrysiasis) and penicillamine can be associated with the development of skin laxity and fragility.22–25
Drug toxicity
Drug toxicity develops as a consequence of the gradual accumulation of a drug or its metabolite (e.g., minocycline or amiodarone deposition with resultant abnormal pigmentation).15,26–29 Delayed toxicity may take months to many years before expression (e.g., arsenical keratoses).30–33
Drug interactions
Drug interactions develop when one drug alters the pharmacological efficacy of another that is given concurrently.12,13,34,35 The effect may enhance or diminish the effect of the drug with resultant toxicity or loss of therapeutic value.13,14 Drug interactions are thought to arise when one drug affects clearance of the other as a consequence of several mechanisms including:3,4
The last is believed to be of particular importance and includes increased enzyme synthesis with excessive drug degradation and diminished circulating or tissue levels and inhibition of drug breakdown with increased circulating and tissue levels.34,36
Drug interactions are of particular importance in the elderly, the immunosuppressed, and in those patients receiving multiple medications.3,4
Type B drug reactions
Type B reactions are uncommon and unpredictable. They do not have an allergic pathogenesis and include: 1
Idiosyncratic drug reactions
Idiosyncratic reactions (drug intolerance) develop as a result of genetic or metabolic influences. They may represent the effects of abnormal or altered hepatic drug metabolism. For example, a lupus erythematosus-like condition is a rare complication of hydralazine therapy in the average population but the risk is greatly increased in patients who metabolize the drug slowly.3,7 Drug-induced lupus erythematosus may also be caused by procainamide, chlorpromazine, isoniazid, methyldopa, penicillamine, minocycline, quinidine, and sulfasalazine.36–38 Cefaclor-induced serum sickness-like eruptions and the antiepileptic hypersensitivity syndrome are also believed to result from reactive intermediate metabolic products.39,40
Exacerbation of a pre-existing condition
This is a not uncommon problem; for example, lithium, beta-blockers, antimalarial drugs, NSAIDs, and tetracycline may precipitate, aggravate or induce a psoriatic eruption.4,41–45
Pseudoallergic drug reactions
Pseudoallergic reactions result from the nonimmunologically mediated release of effector substances such as histamine from tissue-bound mast cells or circulating basophils with resultant urticarial reactions, angioneurotic edema, and anaphylaxis.1,4 The complement system can also be activated by similar nonimmune mechanisms, and there is evidence that perturbation of arachidonic acid metabolism may be involved in some cases.1,3,17 Drugs which have been incriminated in such pseudoallergic reactions include radiocontrast media, NSAIDs, acetyl salicylic acid, opium derivatives, codeine, curare, d-tubocurare, polymyxin B, and angiotensin-converting enzyme (ACE) inhibitors.46–50
Type C drug reactions
Type C reactions are rare, immunologically mediated, and develop as a consequence of previous exposure to the drug with resultant allergy.1 The majority of drugs are of low molecular weight (less than 1000 Daltons) and therefore on their own are incapable of eliciting an immune response. By functioning as haptens and forming conjugates with carrier plasma proteins or cell membrane constituents they develop immunogenic potential.1,2,17 The ability of the majority of drugs to cause an immune response is therefore dependent on whether it is able to bind to circulating or tissue protein.51 A number of drugs are likely to induce allergic reactions, including antibiotics, anticonvulsants, chemotherapeutic agents, heparin, insulin, protamine, and biological response modifiers such as interferons and growth factors.1 A variety of mechanisms may be involved in cutaneous drug-induced hypersensitivity reactions including:12
IgE-mediated type 1 cutaneous reactions
In type 1 reactions, the release of histamine and other chemical mediators from tissue-fixed mast cells results in increased vascular permeability with development of edema in the dermis or deeper tissues.5,17 Immediate reactions which develop within an hour or less of drug exposure present as urticaria, angioedema or anaphylaxis whereas accelerated reactions which develop from 1 to 72 hours following the administration of the drug are usually urticarial.17 Urticaria following treatment with penicillin is a typical type 1 reaction. Certain other antibiotics, antisera, and gammaglobulin are also common offenders.12
The most common cause of anaphylaxis is penicillin.52 Other causes include foods, stings, anesthetics, muscle relaxants, latex, contrast material, antibiotics, and allergenic extracts.52–54 In addition to histamine, anaphylaxis is mediated through a number of substances including prostaglandin D2, leukotriene C4, interleukin (IL)-4 and IL-13, and tumor necrosis factor alpha (TNF-α).51
Immune complex-associated type 3 reactions
Type 3 reactions are expressed as urticaria, the Arthus reaction, serum sickness, and leukocytoclastic (allergic) vasculitis.12 The disease manifests a week or more after exposure to the drug, by which time sufficient circulating antibody has been generated to result in immune complexes of an appropriate size to avoid phagocytosis. Their deposition in the tissues or within blood vessel walls is accompanied by complement fixation and resultant acute inflammatory reaction.
Delayed hypersensitivity type 4 reactions
Delayed hypersensitivity reactions are T-lymphocyte mediated and exemplified in acute allergic contact dermatitis.12,17 Cytotoxic T-cell-mediated reactions are of importance in many other adverse allergic drug reactions including exanthematous/morbilliform, bullous, and interface variants.55–58 Although most delayed hypersensitivity reactions are immune responses to the hapten-carrier complex, recent studies indicate that some drugs may be capable of directly activating the immune system, independent of a covalent drug–peptide complex.59 Certain medications may directly bind T-cell receptors and MHC molecules and trigger the release of cytokines which recruit specific leukocytes. The delayed hypersensitivity reactions may be subclassified based on the cell type recruited: monocytes (type 4a), eosinophils (type 4b), T cells (type 4c), and neutrophils (type IVd). The resultant clinical phenotype may be determined by which cells are involved.59
Adverse drug reactions – clinical manifestations
Although the range of drugs that may result in adverse drug reactions is extensive, the variety of clinical responses encountered is fairly limited. Many drugs may cause more than one clinical response and any given reaction pattern may result from a wide range of drugs. There are, however, a number of clinicopathological responses that are fairly unique to a particular drug and these are dealt with individually, later in this chapter.1–8
Adverse drug reactions may therefore present with a considerable number of clinical manifestations as outlined in Box 14.1.1,2
Exanthematous reactions
Clinical features
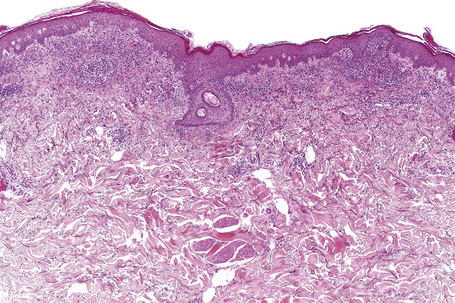
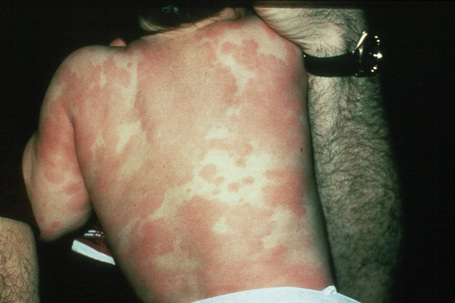
Exanthematous (morbilliform, maculopapular) reactions are the most frequently encountered adverse drug reaction, accounting for 51% to 95% of skin reactions, and mimic a variety of infective conditions including scarlet fever, measles, and rubella (Figs 14.1, 14.2).1–5 Patients present with erythematous macules and papules that may become confluent or gyrate/ polycyclic. Pruritus, low-grade fever, and eosinophilia are sometimes present.2 The eruption is often symmetrical and usually presents on the trunk and extremities or sites of pressure and trauma.1 The palms and soles are sometimes affected but the mucous membranes are not usually involved.
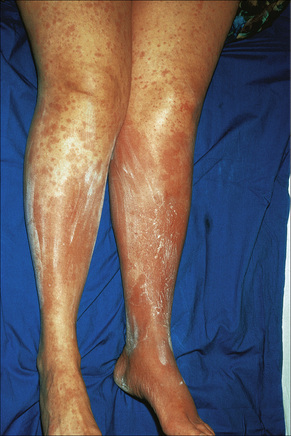
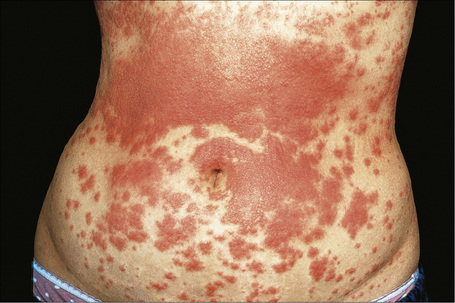
Exanthematous eruptions typically develop within 1–2 weeks of starting the drug.1 Occasionally, the eruption is delayed and may even present after the treatment has ceased.1,6 In more seriously affected patients the eruption can progress to erythroderma (exfoliative dermatitis) in which the erythema becomes generalized and is often accompanied by scaling.7 Resolution of exanthematous drug reactions is characterized by exfoliation and sometimes is followed by postinflammatory hyper- or hypopigmentation.1 Penicillin, sulfonamides, ampicillin, amoxicillin, phenylbutazone, isoniazid, barbiturates, phenytoin, carbamazepine, benzodiazepines, gold, and trimethoprim are especially incriminated.1,8–10 Patients who suffer from infectious mononucleosis are at risk of developing an exanthematous reaction following therapy with ampicillin or amoxicillin.11
Pathogenesis and histological features
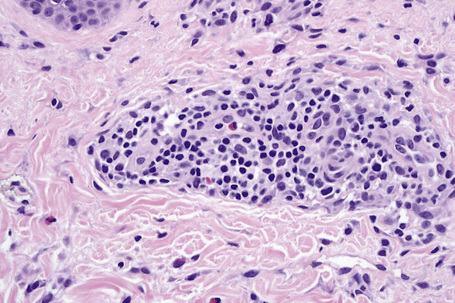
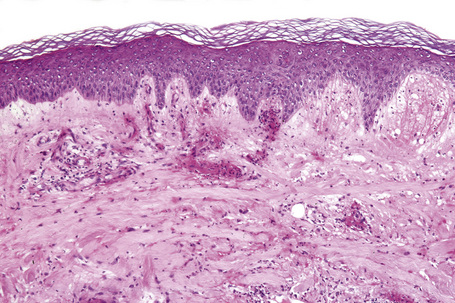
The histological features are often subtle. Although the epidermis may appear normal, focal parakeratosis is commonly present. The characteristic changes include lymphocytic exocytosis with mild spongiosis, typically accompanied by basal cell liquefactive degeneration and a few dyskeratotic keratinocytes (Figs 14.3–14.7).12,13 The dermis shows a perivascular infiltrate of lymphocytes and histiocytes with variable numbers of eosinophils. Eosinophils – although often emphasized in the literature as an important feature of drug reactions – can, in our experience, be very scanty or even absent. Sometimes marked edema is seen, particularly if an urticarial element is clinically evident. Red cell extravasation may also be a feature in those lesions that include a purpuric component.
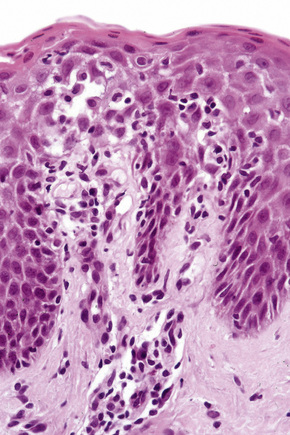
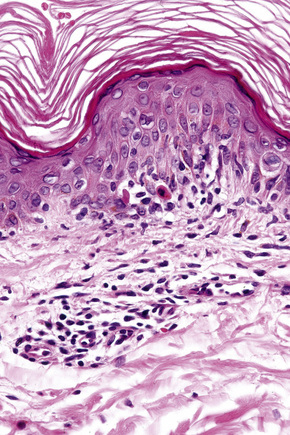
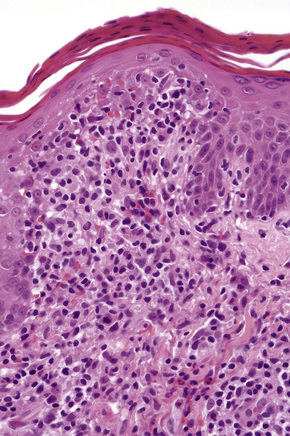
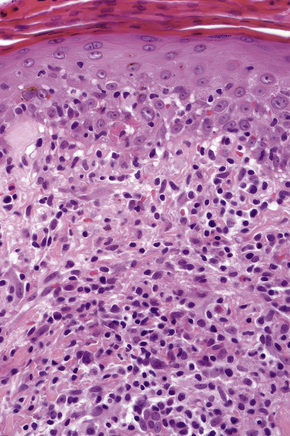
By immunohistochemistry, the lymphocytes are largely CD3+ T-cells with a predominance of CD4+ cells in the superficial perivascular infiltrate.14 Lymphocytes at the dermoepidermal junction and within the epidermis consist of approximately equal numbers of CD4+ and CD8+ forms.14–19 These latter cells regularly express human leukocyte antigen (HLA)-DR and a subpopulation also expresses CD25.19 There is an admixture of T-helper Th1 and Th2 cells.18 Occasionally, the infiltrate is almost entirely composed of the CD4+ lymphocytes and, contrariwise in human immunodeficiency virus (HIV) positive patients, the infiltrate may consist of CD8+ cells alone.14,17 CD1a+ dendritic cells and CD68+ histiocytes are also present.18 CD56+ natural killer (NK) cells may be identified.17 Cytotoxic pathways mediated by perforin and granzyme B have been shown to be of particular importance in exanthematous drug reactions.16,18,19 Fas/Fas-L cytotoxic mechanisms are not thought to be of relevance.14
Differential diagnosis
Exanthematous adverse drug reactions are a frequent feature in transplantation patients who are usually taking multiple medications and, therefore, must be distinguished from acute GVHD. In reality, it is difficult, if not impossible, to make this distinction histologically. Traditionally, the presence of eosinophils has been thought to support a drug reaction. However, a recent study found tissue eosinophils in both graft-versus-host disease and drug reactions, making histological distinction impossible.20 A viral exanthem also commonly enters the differential diagnosis – the histological findings are often indistinguishable although the presence of eosinophils may favor a drug reaction.
Urticarial reactions, angioedema and anaphylaxis
Clinical features
Urticaria is the second most common adverse drug reaction.1 It is characterized by pruritic, erythematous, and edematous wheals. If accompanied by marked edema involving the deeper dermis and subcutaneous fat, or if the submucosal layers are affected, angioedema results.2–4 Urticaria can also be a manifestation of serum sickness and anaphylaxis.
Urticarial reactions may be caused by a large number of drugs. Aspirin, penicillin, ACE inhibitors, and blood products are particularly incriminated.5,6 Drugs which directly stimulate mast cell release of vasoactive substances such as histamine can cause urticaria, include opiates, curare, vancomycin, and polymyxin B.3,7,8 Radiocontrast media may have a similar effect.3 Urticarial reactions due to aspirin and NSAIDs are thought to sometimes be a result of abnormal arachidonic acid metabolism.3
Pathogenesis and histological features
The pathogenesis of urticarial drug reactions includes IgE-mediated type 1 reactions, immune complex mechanisms, and pseudoallergic phenomena (non IgE-mediated).5,9,10
Histologically, urticaria is characterized by dermal edema and vascular dilatation accompanied by a perivascular infiltrate consisting of lymphocytes and eosinophils (Fig. 14.8). Edema is often difficult to appreciate histologically but may be inferred by separation of collagen bundles. Mast cell degranulation may be present.3 Vasculitis is not a feature.
Angioedema is characterized by edema extending into the deeper dermis and subcutaneous fat.
Serum sickness/serum sickness-like drug reactions
Clinical features
Serum sickness develops within 1–3 weeks after taking the serum or vaccine.1–8 It presents with an erythematous maculopapular or urticarial response or with palpable purpura variably accompanied by fever, arthralgia, myalgia, arthritis, lymphadenopathy, glomerulonephritis, myocarditis, and neuritis (Fig. 14.9).1–4 The cutaneous manifestations often commence on the sides of the fingers, toes, and hands before becoming more generalized.2 A wide range of drugs has been implicated in the development of serum sickness-like drug reactions including phenytoin, phenylbutazone, and carbamazepine. Antibiotics are also a common offender, with cefaclor featured most prominently along with other antibiotics such as cefprozil, ciprofloxacin, minocycline, penicillin V, amoxicillin, flucloxacillin, and co-trimoxazole.6,9–19 More recently, therapy with monoclonal antibodies such as rituximab, infliximab, and natalizumab has been associated with serum sickness-like reactions.20–22
Pathogenesis and histological features
Serum sickness is thought to represent an immune complex-mediated type 3 reaction although the possibility of direct toxicity against vessel wall, autoimmunity, and cell-mediated cytotoxicity have been proposed as alternative pathogenetic mechanisms.2 Direct immunofluorescence reveals immunoglobulin and C3 in relation to blood vessel walls.6
The histological features are those of leukocytoclastic vasculitis (Fig. 14.10).23
Phototoxic and photoallergic reactions
Clinical features
There are two types of photosensitive drug reactions: phototoxic and photoallergic. Phototoxic reactions are more common; however, they are not necessarily mutually exclusive and are not always clinically distinguishable.1–30 The clinical appearances of acute phototoxic reactions mimic severe sunburn and include erythema, edema, and blistering with subsequent desquamation and postinflammatory hyperpigmentation (Figs 14.11, 14.12).6,7 Typically, only exposed skin is affected and it occurs minutes to hours after sun exposure. Phototoxicity has also been associated with onycholysis.21,25
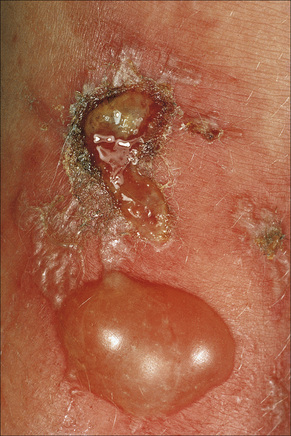
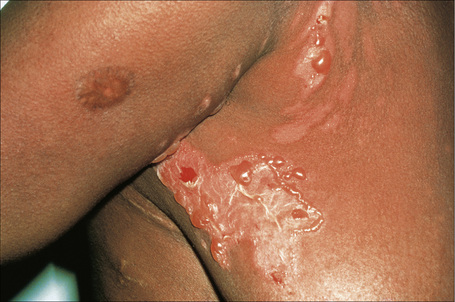
Fig. 14.12 Phototoxic drug reaction: the lesions in this patient followed PUVA therapy.
By courtesy of the Institute of Dermatology, London, UK.
Chronic phototoxicity presents with poikilodermatous features including hyper- and hypopigmentation, epidermal atrophy, and telangiectasia. It is an important feature of the porphyrias and the inherited photodermatoses such as xeroderma pigmentosum, Rothmund-Thomson syndrome, and Bloom’s syndrome. It rarely results from drug treatment but may follow long-term therapy with psoralen and UVA (PUVA therapy) where there is also an increased incidence of actinic keratosis, basal cell carcinoma, squamous cell carcinoma and, more rarely, melanoma.6,31–34
Drugs that are incriminated in acute phototoxic reactions include thiazide diuretics, sulfonamides, tetracycline antibiotics, NSAIDs (including naproxen, diclofenac, and ketoprofen), phenothiazines (particularly chlorpromazine), amiodarone, tars, and psoralens.6–13,19,20,23,24,29 Phototoxicity has also been described with use of St. John’s wort and following photodynamic therapy.22,26 Porphyrins are potent phototoxic sensitizers.6 In this instance, the damage affects the dermal constituents including the vasculature, leaving the epidermis relatively unaffected.
The clinical appearances of photoallergic drug reactions are variable and include eczematous and lichenoid dermatitides (Figs 14.13, 14.14).14 The rash usually develops 24 hours or more after sun exposure. Unlike phototoxic reactions, unexposed skin may also be affected in addition to exposed skin.7 Typically, the dermatitis resolves after withdrawal of the offending agent. Rarely, a persistent light reaction may occur in which the photodermatitis persists despite removal of the photosensitizing chemical. This usually occurs in the setting of photoallergic contact dermatitis.
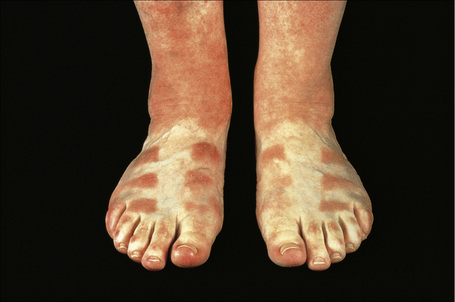
Fig. 14.13 Photoallergic drug reaction: note the obvious sparing of covered skin.
By courtesy of the Institute of Dermatology, London, UK.
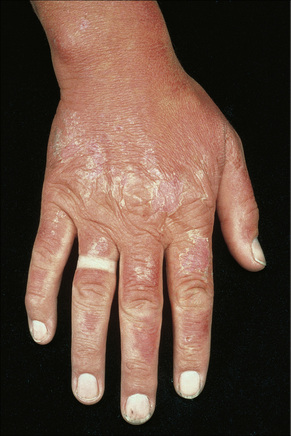
Fig. 14.14 Photoallergic drug reaction: this example resulted from treatment with tetracycline.
By courtesy of the Institute of Dermatology, London, UK.
The majority of photoallergic reactions are induced by the application of topical medicaments and chemicals (contact photoallergy) including antihistamines, local anesthetics, chlorpromazine, hydrocortisone sunscreens containing p-aminobenzoic acid, and halogenated phenolic compounds in soaps and fragrances (e.g., 6-methylcoumarin and musk ambrette).2,7,15,17 Photoallergy can also follow systemic administration of drugs including sulfonamides, griseofulvin, phenothiazines, tetracyclines, NSAIDs, chloroquine, and thiazides.8,18 Diagnosis is best confirmed by a photopatch test.
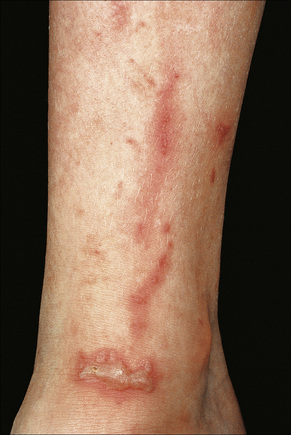
Phytophotodermatitis represents a phototoxic drug reaction due to contact with plants containing furanocoumarins.35–37 Patients develop erythema followed by postinflammatory hyperpigmentation. Rarely, blisters may develop (Fig. 14.15).35 Members of the Umbelliferae, Rutaceae and Moraceae families are implicated.36,37
Pathogenesis and histological features
Photosensitization has been described as a process whereby ‘a reaction to non-ionizing radiation occurs as a consequence of the introduction of a radiation-absorbing reagent (the sensitizer), which induces another substance (the substrate) to undergo chemical change’.1–6 There are two basic mechanisms: phototoxic and photoallergic. While an enormous range of drugs has been implicated in photosensitivity reactions, NSAIDS, phenothiazines, amiodarone, antibiotics, and antifungal agents such as griseofulvin appear to be of particular importance.30
Two types of phototoxic reactions are recognized: photodynamic and non-photodynamic.6
Many drug reactions are photodynamic, whereas psoralen represents a nonphotodynamic reaction. Phototoxic reactions do not depend on prior exposure to the drug and will affect all patients of the same skin type provided that sufficient bound drug is available for reaction with the appropriate radiation.1 The action spectrum for phototoxicity is UVA, and less often UVB and visible light.
Photoallergic drug-induced photosensitivity is immunologically mediated and represents a delayed papular, vesicular or eczematous response.6 It is a lymphocyte-mediated delayed hypersensitivity type 4 reaction. It typically requires previous exposure to the drug or chemical.1,5 Only a proportion of patients taking the drug will develop a reaction. Photoallergic reactions are usually induced by UVA.1,2
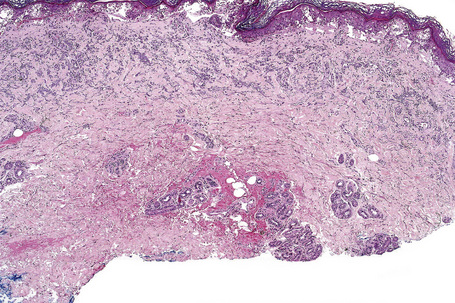
The histological appearances of acute phototoxic reactions include conspicuous apoptotic keratinocytes (sunburn cells) which in severe cases may affect the entire epidermis, with variable neutrophil exocytosis, dermal edema, and a perivascular lymphohistiocytic infiltrate with small numbers of neutrophils and eosinophils (Figs 14.16, 14.17).38
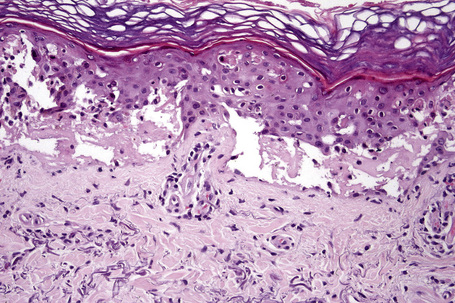
Chronic lesions are characterized by hyperkeratosis, hypergranulosis, variable acanthosis and epidermal atrophy, increased melanin pigmentation, melanocyte hyperplasia, and pigmentary incontinence.38 Elastosis and telangiectatic vessels may be conspicuous, and in severely affected patients stellate atypical myofibroblasts can be a feature. Epidermal disorganization and dyskeratosis may also be present.6
The histological appearances of drug induced photoallergic reactions includes spongiosis (often with vesiculation, lymphocytic, and eosinophil exocytosis), accompanied by papillary dermal edema and an upper dermal lymphohistiocytic infiltrate with variable numbers of eosinophils.38
Anticonvulsant hypersensitivity syndrome
Adverse drug reactions to phenytoin – which include erythematous maculopapular lesions, erythroderma, acneiform lesions, hypo- and hyperpigmentation, vasculitis, erythema multiforme, and toxic epidermal necrolysis – affect up to 19% of patients taking this drug.1–7 Pseudolymphomatous drug reactions may also occur and these are discussed later in the chapter.
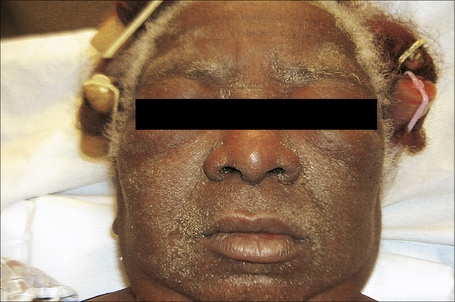
A potentially fatal hypersensitivity syndrome develops in approximately 1 in 3000 patients receiving phenytoin.5,6 This is defined by a triad of pyrexia, exanthematous skin rash, and evidence of systemic involvement.6 The acronym DRESS describes drug rash with eosinophilia and systemic symptoms. Administration of other antiepileptics including phenobarbital, carbamazepine, primidone, and lamotrigine may result in an identical condition.8–11 It can also be caused by other medications including allopurinol, azathioprine, dapsone, minocycline, sulfonamides, terbinafine, and more recently efalizumab.12–17 The syndrome has been predominantly described in black patients. There is no sex predilection.4 Children may be affected.18–22 Symptoms appear 1 to 8 weeks after starting the offending drug. Clinical features include pyrexia, a maculopapular or erythrodermatous eruption, facial or periorbital edema, strawberry tongue, tender lymphadenopathy, myositis and hepatitis associated with leukocytosis and eosinophilia (Figs 14.18, 14.19).4,5 Less often the cutaneous manifestations include localized or generalized follicular pustules, erythema multiforme, and toxic epidermal necrolysis.4,6,22,23 In patients with the pustular variant, lesions present on the scalp before becoming generalized.8,23,24 Conjunctivitis and/or pharyngitis may also be present.2 Renal, pulmonary (interstitial pneumonitis), and hematological (atypical lymphocytosis) involvement sometimes occur.5 The prognosis of this syndrome is variable.25,26 The majority of patients recover but in those with hepatitis, the mortality is approximately 20%.5,27

Pathogenesis and histological features
The precise etiology of this syndrome is uncertain. It is thought to result from an inability to detoxify arene oxide anticonvulsant metabolites due to absence, possibly genetically determined, of specific hydrolases. The condition has been associated with reactivation of human herpes virus 6 and 7, Epstein-Barr virus, and cytomegalovirus.28–33
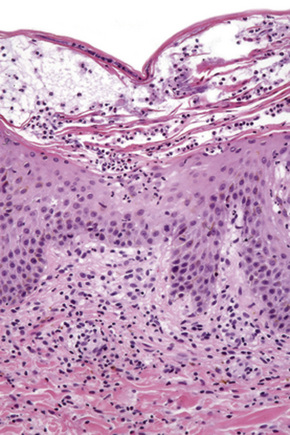
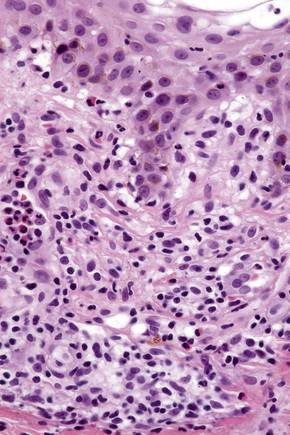
The histological features vary from spongiotic dermatitis to those of erythema multiforme or toxic epidermal necrolysis. Pustular lesions are characterized by a subcorneal pustule associated with follicular infundibular dilatation (Figs 14.20, 14.21).8
Lichenoid and interface drug reactions
Clinical features
Lichenoid drug reactions are clinically similar to lichen planus although lesions are often larger, Wickham’s striae are usually not apparent, and mucosal involvement is commonly absent.1–4 In contrast to lichen planus, where lesions are characteristically on the flexural surfaces of the forearms, the legs, and the genitalia, in lichenoid drug reactions the trunk and extremities are more often affected (Figs 14.22, 14.23 ).4,5 The eruption may sometimes be photodistributed and predominantly affect the hands and forearms although other sun-exposed sites can be involved.4,6,7 The latent period between starting the drug and the onset of the eruption is often long, months or even years.4 Atypical features including eczematous and psoriasiform lesions are sometimes seen and bullous or ulcerative variants are occasionally encountered.3,4,8 Postinflammatory hyperpigmentation may be very marked and is often persistent. Scarring alopecia is sometimes present and some patients may develop anhidrosis.4
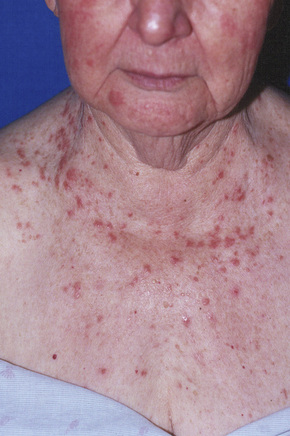
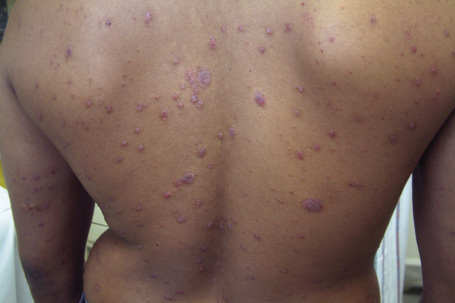
Fig. 14.23 Lichenoid drug reaction: lichenoid papules on the back.
By courtesy of B Al-Mahmoud, MD, Doha, Qatar.
Although many drugs can cause a lichenoid reaction, those of particularly importance include gold, antimalarials such as quinine and quinidine, penicillamine, captopril, various beta-blockers (e.g., propranolol), lithium, thiazide diuretics, furosemide (frusemide),spironolactone, and ethambutol.4,7–19 More recently, the TNF-alpha inhibitors infliximab, adalimumab, and etanercept have been associated with lichenoid eruptions.20–22
Other causes of a contact lichenoid eruption include dental restorative materials, musk ambrette, nickel, and gold.4
Captopril and cinnarizine may cause lichen planus pemphigoides-like eruptions (see bullous drug reactions below).23,24
Histological features
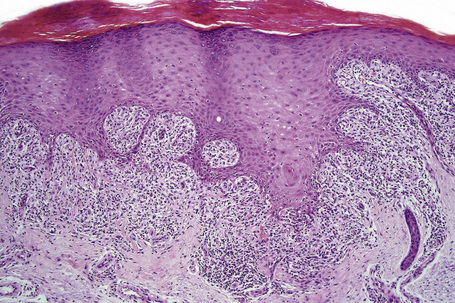
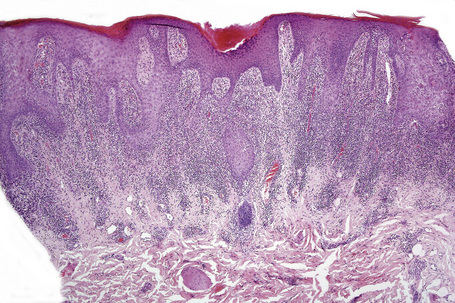
The histological features are frequently indistinguishable from typical lichen planus although focal parakeratosis and spongiosis are sometimes present and interface change may be patchy (Figs 14.24–14.26). The epidermis is often thinner and hypergranulosis less marked.15 Cytoid bodies may be found in the upper granular cell layer or even in the stratum corneum.5,7 Sometimes, eosinophils and occasionally plasma cells are found in the dermal infiltrate.5 Focal interruption of the granular cell layer, exocytosis of lymphoid cells into the upper epidermis and a perivascular infiltrate in the deeper dermis are said to be additional helpful diagnostic pointers.7 Photodistributed lichenoid drug reactions are said to more closely resemble idiopathic lichen planus than nonphotodistributed variants.5 The changes that allow distinction from lichen planus are often seen in hypertrophic lichen planus. However, in lichenoid drug eruptions, prominent hyperplasia is hardly ever present. With some drug reactions, interface changes are present in the absence of a background of epidermal lichenoid features (Figs 14.27, 14.28).
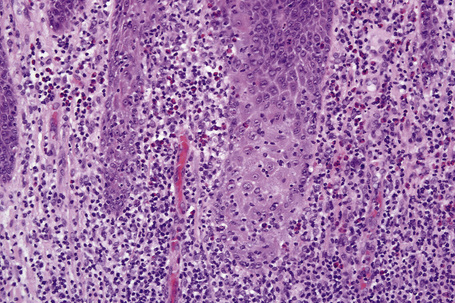
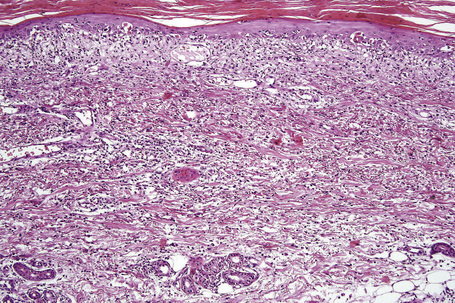
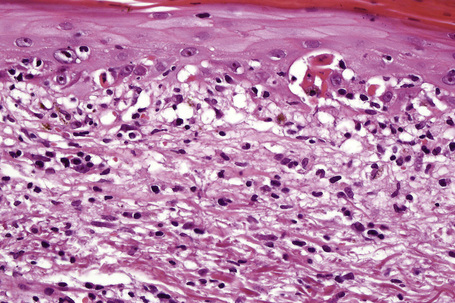
Fixed drug eruptions
Clinical features
Fixed drug eruptions present as one or more circumscribed erythematous to violaceous or brown plaques that show a predilection for the extremities including the hands, feet, and external genitalia (Figs 14.29, 14.30).1–8The mucous membranes may be affected, either alone or in association with cutaneous manifestations.3 Lesions – which may be pruritic or present with a burning sensation – typically recur at the same site on rechallenge with the offending drug. They usually develop within 30 minutes to 8 hours after taking the drug.3 Vesiculation and blistering are common. Resolution is typically marked by postinflammatory hyperpigmentation varying from brown to brown-violet or even black.6 In cases with multiple lesions the multiple pigmented patches result in an appearance described as ‘Dalmatian dog’. Occasionally, the eruption is generalized and resembles toxic epidermal necrolysis.9 Although the number of drugs that are capable of eliciting a fixed reaction is very large, those that are said to be more commonly incriminated include barbiturates, phenylbutazone, ibuprofen, acetyl salicylic acid, sulfonamides, trimethoprim-sulfamethoxazole, tetracyclines, dapsone, phenolphthalein, and quinine.2,3,6
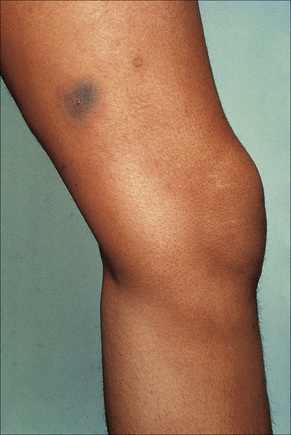
Fig. 14.29 Fixed drug eruption: typical localized brown plaque with a small central blister.
By courtesy of the Institute of Dermatology, London, UK.
Pathogenesis and histological features
Fixed drug eruption is unique owing to the precise localization of the eruption and its recurrence at the same site on rechallenge. To understand this process, initial research was directed towards identifying the site of cutaneous memory. Autotransplantation experiments in which normal skin was grafted to a previously affected site and vice versa, followed by rechallenge with the causative drug, produced conflicting results. Some workers found that following challenge, grafted normal skin was unaffected, whereas transplanted previously affected skin developed erythema and became symptomatic.10 Others experienced quite the opposite results.11
Immunofluorescence studies have been equally conflicting. While some authors have documented in vivo bound immunoglobulin and complement in the intercellular region of the epidermis or at its basement membrane, the majority of investigations have been negative.12,13
It seems unlikely, therefore, that humoral immunity has a significant part to play in the pathogenesis of fixed drug eruption. Current research is directed towards understanding the role of cellular immunity. On initial exposure, the drug appears to bind to the epidermal keratinocytes (thereby functioning as a hapten) and is presented by Langerhans cells to lymphocytes within the dermis or in local lymph nodes. This stimulates an effector CD8+ lymphocyte population which, on returning to the epidermis, produces various cytokines including interferon-gamma (IFN-γ) and TNF-α which result in epidermal necrosis.14,15 Keratinocyte death is believed to be mediated by both cytolytic pathways (e.g., perforin, granzyme A, and granzyme B) and FAS-mediated apoptosis.14–16 Resolution of the disease is thought to be due to recruitment of CD4+ T cells into the epidermis which suppress CD8+ T-cell activation and limit cell destruction, possibly via production of IL-10.17
Although the precise mechanism by which memory in fixed drug eruption is achieved is incompletely understood, there is now considerable evidence to suggest that an intraepidermal effector-memory CD8+ T-cell population residing in the epidermis after the initial drug reaction is of particular importance.13–20 Such cells are defined immunohistochemically by expression of CD3, CD45RA, TCR-alpha beta, CD11a, and CD11b, and absence of CD27, CD28, and CD62L.19,21 It has been demonstrated that they remain in a state of activation (CD69+) in the unchallenged state and, following exposure to the drug, rapidly up-regulate IFN-γ expression and induce FAS and FAS-ligand expression, soon followed by epidermal necrosis.16,20
Histologically, the acute fixed drug eruption is characterized by marked basal cell hydropic degeneration, with lymphocyte tagging along the dermoepidermal junction and individual keratinocyte necrosis (Figs 14.31–14.33).22 Marked pigmentary incontinence is typical. Subepidermal vesiculation may be a feature of advanced lesions. Lymphocytes, histiocytes, and neutrophils are evident in the upper dermis. Eosinophils may sometimes be prominent. In late lesions, pigmentary incontinence may be the sole histological finding.
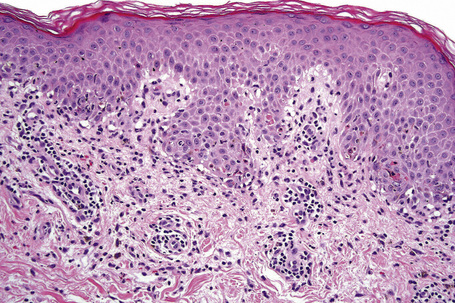
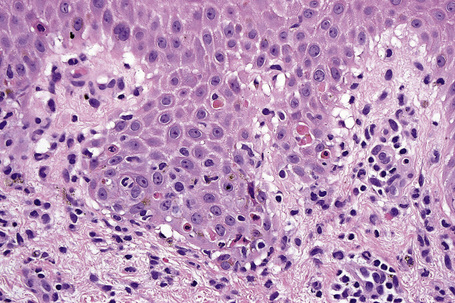
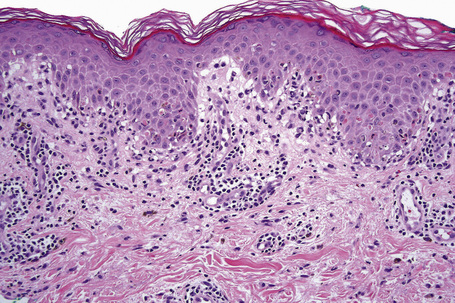
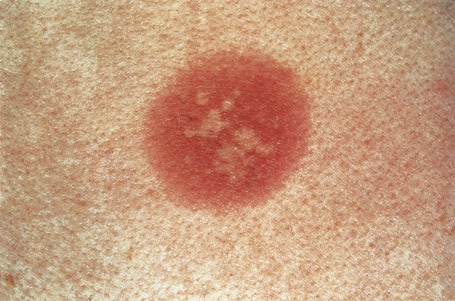
Erythema multiforme
Although infectious agents (herpes simplex virus, Mycoplasma species) are the most common cause of erythema multiforme (EM), medications, or a combination of medications and viral infections, are implicated in a subset of patients. Drugs with the strongest association include antibiotics, anticonvulsants, and nonsteroidal antiinflammatory agents.1 Sulfonamides, specifically trimethoprim-sulfamethoxazole, carry the highest relative risk. Other causative antibiotics include aminopenicillins, quinolones, cephalosporins, and tetracyclines. The anticonvulsants associated with EM most often are phenobarbital, carbamazepine, phenytoin, and valproic acid. EM due to a combined viral infections and drug exposure has been described with cytomegalovirus and Epstein-Barr virus.2,3
Stevens-Johnson syndrome and toxic epidermal necrolysis
In contrast to erythema multiforme, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are strongly associated with medication use.1 The profile of implicated drugs is similar to that seen in EM (see above).2 Studies on newer medications cite strong associations for nevirapine, lamotrigine and weaker but significant associations for sertraline, pantoprazole and tramadol.3
Drug-induced hyperpigmentation
Clinical features
Cutaneous hyperpigmentation is a frequent complication of drug therapy. It may result from increased melanin synthesis or deposition of the drug or its metabolite within the skin.1–5 Heavy metals can also result in skin pigmentation.4 Most often, however, it results from postinflammatory hyperpigmentation.3
Long-term treatment with minocycline might result in usually reversible (types I and II) cutaneous pigmentation.6–13 Three clinical variants of cutaneous minocycline pigmentation are generally recognized (Figs 14.34–14.36):
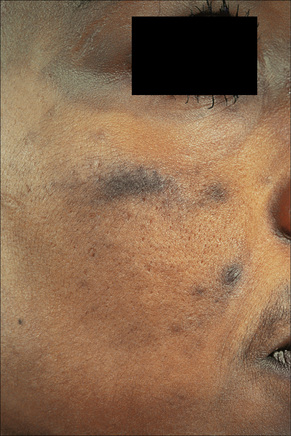
Fig. 14.34 Minocycline pigmentation: extensive lesions involving the cheek and periorbital region.
By courtesy of the Institute of Dermatology, London, UK.
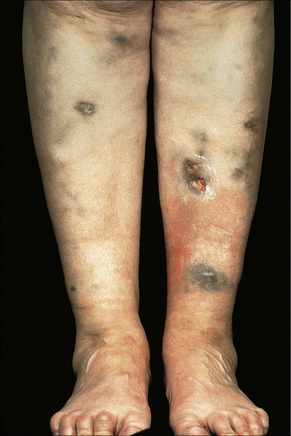
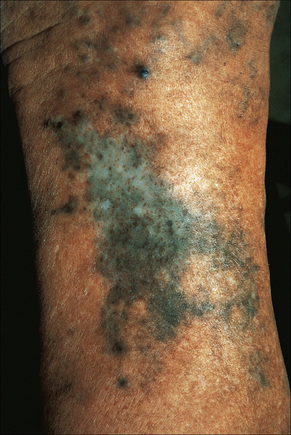
Fig. 14.36 Minocycline pigmentation; typical pigmentation affecting the shin.
By courtesy of the Institute of Dermatology, London, UK.
A fourth variant affecting the lips and possibly representing a fixed drug eruption has been described.14
Nail pigmentation most often presents as a persistent slate-gray coloration of the proximal nail bed.15 Additional features include longitudinal melanonychia, diffuse nail pigmentation, and photo-onycholysis.15
Minocycline may involve the teeth (causing a green–gray or blue–gray discoloration) predominantly affecting the middle and occasionally the incisal thirds of the crown.16 Lesions of the oral mucosa are rare although pigmentation has been described on the buccal mucosa, gingiva, tongue, and lips.17–21 The bones underlying the oral cavity (black bone disease) represent the single site most commonly affected by minocycline pigmentation.22 This is best visualized by inspecting the maxillary and mandibular anterior alveolar mucosa.14 The hard palate and lingual alveolar bone are also commonly affected.14
The conjunctiva, sclera, thyroid (black thyroid), aorta, endocardium, and atherosclerotic plaques may also be involved in minocycline pigmentation.23–29
Many other tetracyclines including methacycline and tetracycline hydrochloride have also been associated with cutaneous pigmentation.30,31
Amiodarone, which is used primarily in the treatment of cardiac arrhythmias, is associated with a phototoxic/photosensitivity reaction in up to 50% of patients.32–38 In addition, cutaneous golden-brown to slate-gray or blue/violaceous pigmentation predominantly affecting the exposed surfaces including the face and the backs of the hands may develop, especially in those receiving high doses over a protracted period of time (Fig. 14.37).32 Pigmentation is also sometimes seen in the sclera and cornea.35
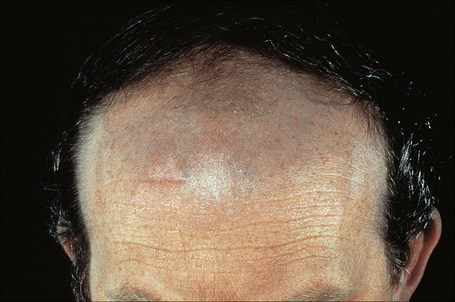
Fig. 14.37 Amiodarone pigmentation: note the slate-gray discoloration on the forehead, a characteristic site.
By courtesy of the Institute of Dermatology, London, UK.
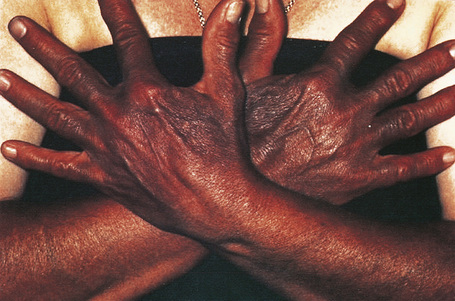
Antimalarials also result in abnormal skin pigmentation.39–42 Mepacrine (quinacrine) typically produces a yellow coloration although localized blue–black mucocutaneous lesions have been described (Figs 14.38, 14.39).42 Chloroquine and hydroxychloroquine cause yellow–brown to gray pigmentation.2,39–41 Sun-exposed skin is predominantly affected, although mucosal pigmentation may also occur.43
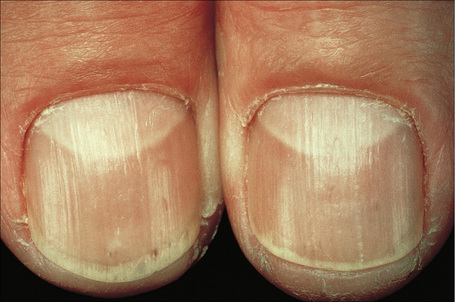
Fig. 14.38 Mepacrine pigmentation: a yellow discoloration is characteristic.
By courtesy of the Institute of Dermatology, London, UK.
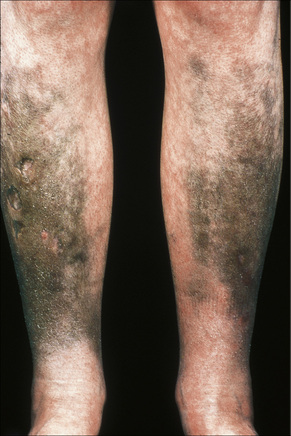
Fig. 14.39 Mepacrine pigmentation: in this patient the drug resulted in black lesions.
By courtesy of the Institute of Dermatology, London, UK.
In addition to causing photosensitivity and contact dermatitis, chlorpromazine therapy (particularly when protracted and in high doses) can result in cutaneous pigmentation, especially on sun-exposed skin such as the face, dorsum of the hands, and the neck.2,44–47 Patients may present with a golden-brown, tanned appearance while others develop a slate-gray, bluish or purple appearance. The cornea and lens of the eye can also be involved.2
Long-term treatment with imipramine may result in photodistributed hyperpigmentation affecting the face, neck, ‘V’ of chest, arms, and hands (Fig. 14.40).48–50 The coloration varies from golden-brown to blue-gray or slate-gray. The irises may also darken.
Photodistributed blue-gray pigmentation has been documented following treatment with desipramine.51
Pathogenesis and histological features
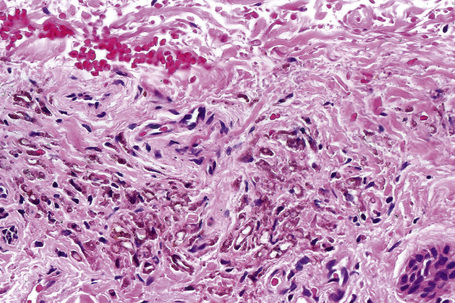
The histological features of minocycline pigmentation are variable.7,10,11,14 In types I and II variants, golden-brown to brown–black granules are found predominantly within macrophages distributed mainly around the vasculature and sweat gland coils (Fig. 14.41). The pigment, which fluoresces yellow under ultraviolet light, stains positively with both Masson-Fontana and Perl’s Prussian blue reactions in type II variants (Figs 14.42, 14.43).11 The pigment is periodic acid-Schiff (PAS) negative. In contrast, in type I, the pigment only stains with Perl’s reaction. It is believed to represent minocycline or its breakdown product chelated with hemosiderin, ferritin or iron.7 Calcium, sulfur, and chlorine are also present but melanin is absent.11 Melanocytes and the epidermis show no increase in melanin pigmentation in types I and II variants. Type III hyperpigmentation is characterized by an increase in epidermal basal cell melanin pigmentation.6 The Perl’s stain is negative. Minocycline pigmentation of the subcutaneous fat has recently been described in the clinical setting of type II disease.52,53 Histologically, there is pigment within macrophages and giant cells in the subcutaneous fat, with positive staining for Masson-Fontana and variable staining with Perl’s reaction. One study also described green–gray nonrefractile globules within macrophages in the fat.53
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


