Chapter 35 Connective tissue tumors
Introduction
A number of cutaneous mesenchymal tumors have genetic features that can be helpful diagnostically. In general, malignant soft tissue tumors fall into the class of complex karyotype sarcomas (for example, angiosarcoma and leiomyosarcoma) or simple genetic profile (for example, clear cell sarcoma or dermatofibrosarcoma) often associated with a chromosomal translocation or, less often, with mutation or loss of a specific gene. An exhaustive discussion of the molecular diagnostics of soft tissue tumors is beyond the scope of this chapter, but see Chapter 2 for additional discussion of relevant techniques. Most of the molecular features discussed in this present chapter can be used diagnostically when required, although some of the tests are available only in specialized centers.
ADIPOCYTIC TUMORS
Benign adipocytic tumors
Lipoma
Clinical features
Lipomas are the most common connective tissue tumors.1–3 They appear to occur more frequently in the obese, usually in middle and late adult life, may be multiple and are purportedly more common in females. This, however, may only be a reflection of the greater tendency of women to request cosmetic attention for otherwise innocuous lesions. Lipomas are very uncommon in children and when present should raise the possibility of Bannayan-Riley-Ruvalcaba syndrome.4 Congenital lipomas are very rare.5 Multiple lesions may occur in a familial setting.6 A case of multiple lipomas after total body electron beam therapy for mycosis fungoides has been reported.7 Multiple lipomas have also been described in association with rosiglitazone, a peroxisome proliferator-activator receptor (PPAR) gamma agonist, in association with systemic chemotherapy for Hodgkin’s lymphoma and in Cowden’s disease.8–10
The lesions are found most often on the trunk, abdomen or neck, followed by the proximal extremities, and rarely on the face (particularly the forehead), scalp, hands or feet. Palmar lipomas are exceptional and may present with lesions simulating piezogenic pedal papules.11 Periungual and subungual lesions are exceptional.12,13 Trauma has been associated with induction of lipomas although it is not clear whether all of these lesions may represent pseudolipomas.14,15 The latter sometimes includes an intravascular lesion.16 Typically, they originate subcutaneously, are slow growing, mobile and painless; sometimes they are multiple. Size varies and some lesions are very large. Dermal examples are often clinically confused with fibroepithelial polyps. Although the lesions are usually well circumscribed, the less common deep variants, which may arise in muscle or in association with a tendon sheath or nerve, are generally ill defined and infiltrative.
Pathogenesis and histological features
Lipomas at all locations show clonal karyotypic abnormalities in up to 75% of cases.17–20 The most common rearrangement in the 12q13~15 region and the HMGA2 gene most often with LLP at 3q27~28, though other loci are described.17–22
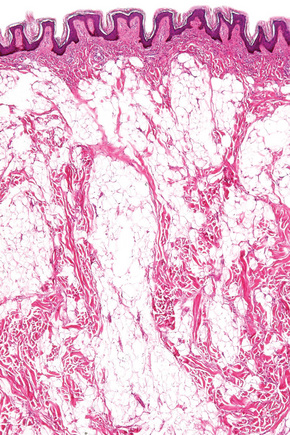
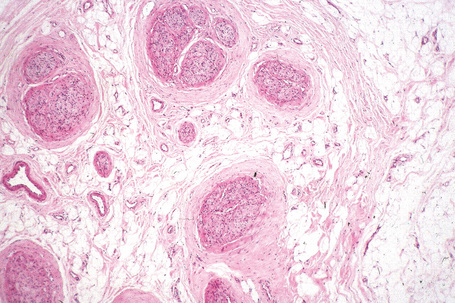
The tumors are usually encapsulated, lobulated and largely composed of univacuolated mature adipocytes, the nucleus and cytoplasm of which are compressed centrifugally (Fig. 35.1). The lobules are divided by delicate fibrous septa containing thin-walled vessels. Degenerative changes, often characterized by fibrosis, focal fat necrosis or myxoid change, are not uncommon, particularly in long-standing or frequently traumatized cases (Figs 35.2–35.00150). Prominent myxoid change and high vascularity are sometime present.23 Foci of other fully differentiated mesenchymal elements, including bone or cartilage, may also be seen.
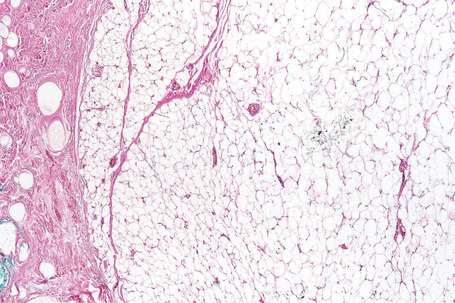
Fig. 35.1 Lipoma: low-power view showing a circumscribed encapsulated tumor composed of mature adipocytes.
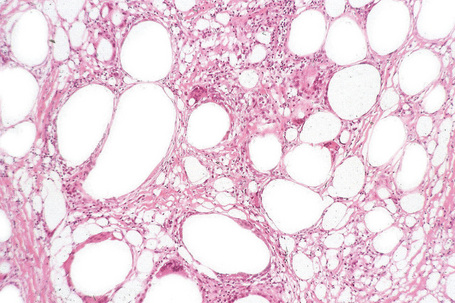
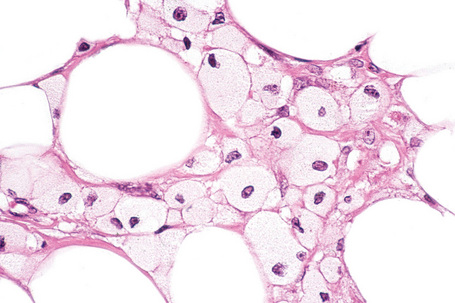
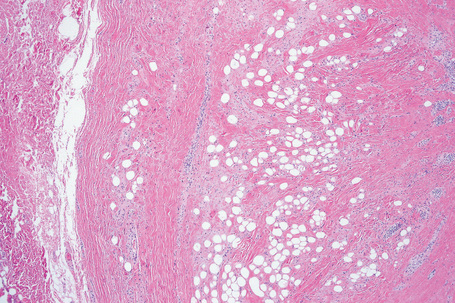
Fig. 35.4 Fibrolipoma: this term is sometimes applied to a lipoma with a prominent fibrous component.
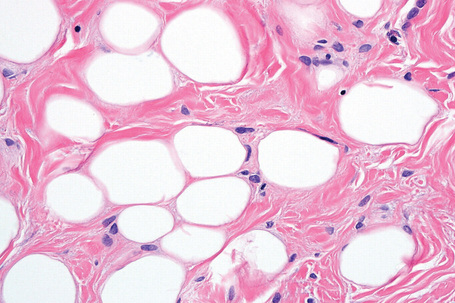
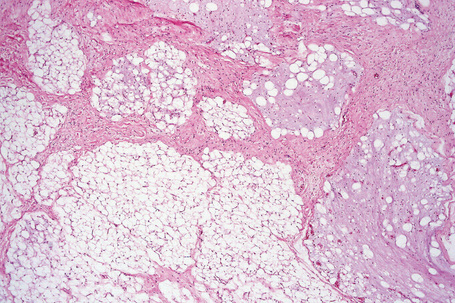
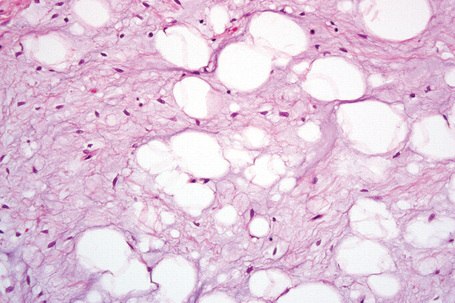
Fig. 35.7 Myxofibrolipoma: note the abundant mucinous matrix and spindled cells admixed with adipocytes.
Although nuclear pleomorphism, hyperchromasia and mitotic activity do not occur in lipomas, in any benign fatty lesion (or even in normal adipose tissue) occasional vacuolated nuclei known as lochkern may be seen (Fig. 35.8). These must not be confused with lipoblasts, the most important diagnostic feature of liposarcoma, which are characterized by multiple intracytoplasmic lipid vacuoles associated with scalloping of peripherally located hyperchromatic or bizarre nuclei. In some lesions there are septa of collagen between adipose tissue lobules and they are referred to as fibrolipomas. A variant of lipoma with predilection for acral sites (fingers, wrists, toes) and characterized by prominent collagenous or myxocollagenous stroma, bland stellate or spindle-shaped cells and scattered adipocytes has been described as sclerotic (fibroma-like) lipoma.24 A further variant known as fibrohistiocytic lipoma has been reported and this is described below.25 Rare lipomas may contain bone.26 Those containing sweat glands do not represent an adenolipoma but rather entrapment of normal glands by the tumor.27–29 Lesions containing smooth muscle are rare; they have been described as myolipomas and tend to be deep seated.30 Focal areas of fat necrosis with foamy histiocytes are often seen and some cases show membranous fat necrosis.31
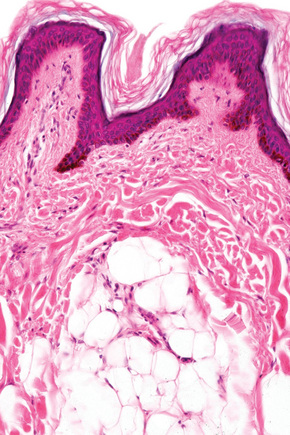
Dermal lipomas are less well circumscribed and consist of scattered groups of mature adipocytes between collagen bundles (Figs. 35.9, 35.10).
Differential diagnosis
The diagnosis of a lipoma is usually straightforward. Distinction from an atypical lipomatous tumor is based on the presence of adipocytes varying in size and shape and with hyperchromatic nuclei in the latter. Dermal lipomas may be confused with pseudolipomatosis cutis. The latter is an artifactual incidental finding distinguished from a dermal lipoma by the presence of empty, round spaces simulating adipocytes but lacking nuclei and of variable size.32
Angiolipoma
Clinical features
Angiolipomas are benign lesions which, in contrast to simple lipomas, are seen most often in young adults and have a predilection for the subcutis of the upper limbs, particularly the forearm and less commonly the trunk.1–3 Oral, intra-articular, extradural, breast and bronchial lesions are exceptional.4–8 Familial cases are rarely seen.
The lesions are typically tender or painful, less than 2 cm in diameter and may impart a reddish or bluish discoloration to the overlying skin. They are more often multiple than solitary and treatment can be problematic. Multiple lesions have exceptionally been documented in association with diabetes mellitus and as a complication of antiretroviral therapy (particularly with indinavir and saquinavir) in acquired immunodeficiency syndrome (AIDS).9–12 A case of intravascular lymphomatosis presenting in an angiolipoma has been documented.13 Metastatic melanoma within an angiolipoma has also been described.14
Pathogenesis and histological features
Cytogenetic studies in angiolipoma have consistently shown a normal karyotype. This is in contrast with other benign lipomatous tumors (including ordinary lipoma) which show characteristic cytogenetic abnormalities.15 This suggests that angiolipomas may have a completely different pathogenesis from that of lipomas.
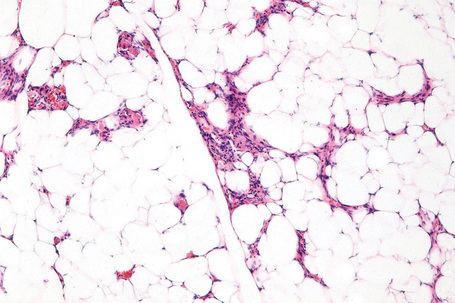
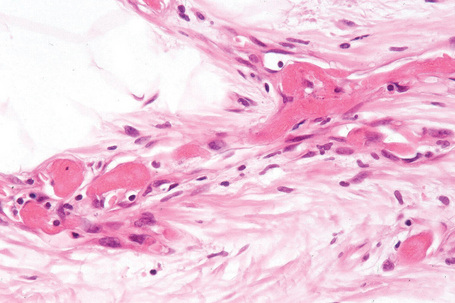
The tumor, almost always encapsulated, is composed of mature adipocytes and varying proportions of irregular, anastomosing small blood vessels without endothelial atypia (Fig. 35.11). Luminal microthrombi are invariably present (Fig. 35.12). Although the blood vessels are usually seen in the periphery of the tumor, they may constitute most of the lesion. Such examples are known as cellular angiolipomas (Figs 35.13, 35.14).16,17
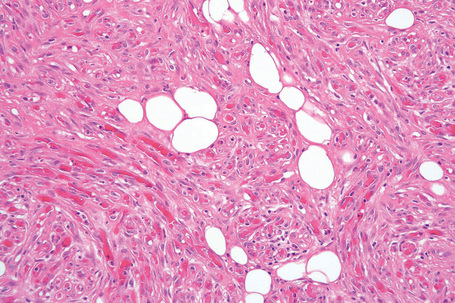
Differential diagnosis
Cellular angiolipoma can be confused with a vascular tumor, especially immature capillary hemangioma and kaposiform hemangioendothelioma.16 The presence of mature adipocytes and capillaries with microthrombi allows distinction from immature capillary hemangioma. Kaposiform hemangioendothelioma may have capillaries with microthrombi in the periphery of tumor lobules, but mature adipocytes are absent.
Spindle cell lipoma
Clinical features
Spindle cell lipoma is a comparatively uncommon variant and may be a source of histological concern to the unwary.1–3 Found predominantly in males, they arise mainly in the posterior portion of the neck, shoulder or upper back, and are characteristically seen in the sixth or seventh decade. Multiple lesions are rare and some are familial.4,5 Rare cases can occur at other locations including the foot, oral cavity (including the tongue), larynx, orbit, soft tissues of the perianal area, aortic valve, mediastinum and breast.6–16 The last may represent an example of mammary-type myofibroblastoma of soft tissue, a lesion closely related to spindle cell lipoma.17 They usually occur as a well-circumscribed, slowly growing, solitary, subcutaneous or (rarely, in up to 13% of cases) dermal lesion, less than 5 cm in diameter.18–20 The last appear most commonly on the face and are more common in females. Intramuscular presentation is very rare.21–24, A case has been reported at the site of an infantile fibrosarcoma treated with chemotherapy.25 Spindle cell lipoma is an entirely benign, nonrecurring lesion.
Pathogenesis and histological features
A number of chromosomal abnormalities have been found in spindle cell lipoma, identical to those found in pleomorphic lipoma. Monosomy or partial loss of chromosomes 13 and 16 are the most common alterations also seen in pleomorphic lipoma, strongly suggesting that these two lesions exist as a morphologic continuum.26–29 Interestingly, similar loss of 13q has been found in mammary-type myofibroblastoma and cellular angiofibroma, suggesting a close link between these tumors.18,30,31
Subcutaneous lesions tend to be well circumscribed and dermal tumors are ill defined.20 In addition to mature univacuolated adipocytes, irregular collections of slender spindled cells are seen with pale eosinophilic cytoplasm, uniform nuclei and rare mitoses (Fig. 35.15). Hyaline bundles of collagen and occasional giant cells may also be present, but lipoblasts are rarely identified. Mast cells are often numerous (Fig. 35.16). The relative proportions of adipose tissue to spindled cells vary between individual cases (Fig. 35.17). Some cases contain few or, rarely, no adipocytes at all.32 Vascularity also varies and focal hemangiopericytoma-like areas are sometimes seen. Extensive myxoid change can lead to striking degenerative features with a pseudovascular pattern in which papillary structures project into empty spaces (Fig. 35.18).33 However, it has been shown that at least in some examples showing this change, the spaces are truly lined by endothelial cells, and these should be named angiomatous spindle cell lipoma.34
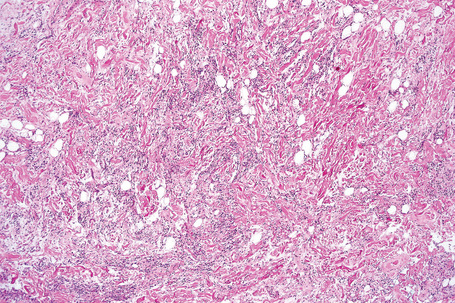
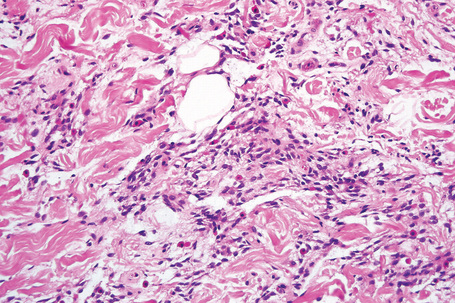
Ultrastructural studies show cells with features of mature adipocytes and spindled cells representing undifferentiated mesenchymal cells.33
Mammary-type myofibroblastoma of soft tissue
Clinical features
This is a rare tumor identical to myofibroblastoma that occurs in the breast.1 As with the latter tumor, it is most common in adult males and presents as an asymptomatic slowly growing subcutaneous mass with predilection for the groin/inguinal area. Size varies but tends to be less than 2 cm. Lesions can also rarely occur on the trunk, vaginal wall, paratesticular area, vulva, perianal area and lower limb.1–5 There seems to be a predilection for tumors to occur along the milk line. Simple excision is the treatment of choice and there is no tendency for local recurrence.
Pathogenesis and histological features
Mammary-type myofibroblastoma appears to be part of a morphologic spectrum that includes cellular angiofibroma and spindle cell lipoma. It has been suggested that the histological variations probably depend on anatomic location.6
Tumors are well-circumscribed and composed of a mixture of bland spindled cells and variable, sometimes prominent, amounts of mature adipose tissue.1 The spindled cells are arranged in fascicles and display a myofibroblast-like appearance with tapering nuclei and amphophilic cytoplasm with an indistinct cytoplasmic membrane. Cytologic atypia is rare and mitotic figures are exceptional. Epithelioid cells may rarely be seen and sometimes scattered multinucleate cells can be identified. Blood vessels are small and tend to be inconspicuous. The stroma is often myxoid, mast cells are common and hyalinized collagen bundles are intermixed with tumor cells.
Immunohistochemistry shows positivity for CD34 and desmin and focal positivity for actin and calponin in a number of cases.1
Cytogenetic analysis has shown partial monosomy of chromosomes 13q and 16q, further emphasizing the relationship with spindle cell lipoma, a tumor with similar cytogenetic abnormalities.7
Pleomorphic lipoma
Clinical features
Pleomorphic lipoma represents a variant of spindled cell lipoma, from which it is clinically and cytogenetically indistinguishable; it is entirely benign.1–5
Pathogenesis and histological features
Cytogenetic abnormalities are the same in pleomorphic lipoma and spindle cell lipoma, confirming that they are part of the same spectrum (see above).4,5 In addition to the histological features of spindle cell lipoma, pleomorphic lipoma is characterized by numerous hyperchromatic and irregular multinucleated giant cells, with nuclei often arranged in a concentric floret pattern (floret giant cells) (Figs 35.19–35.21). Floret-like cells may also be seen in prolapsed orbital fat as a result of a degenerative process.6 Mitotic figures may rarely be evident and occasional multivacuolated lipoblasts are sometimes present (Fig. 35.22). Lesions that show few or no adipocytes may pose a diagnostic challenge.7
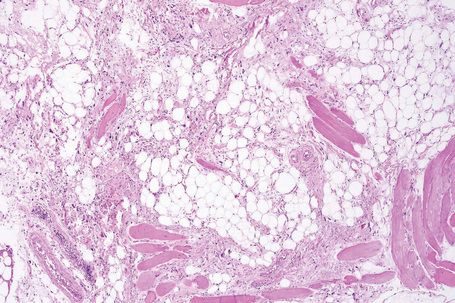
Fig. 35.19 Pleomorphic lipoma: this view shows adipocytes, thick collagen bundles and spindled cells.
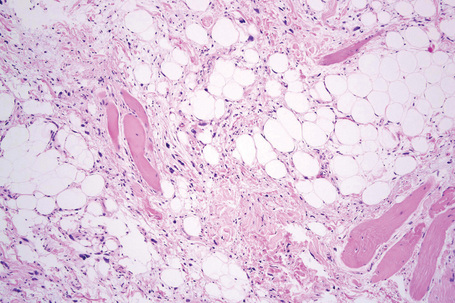
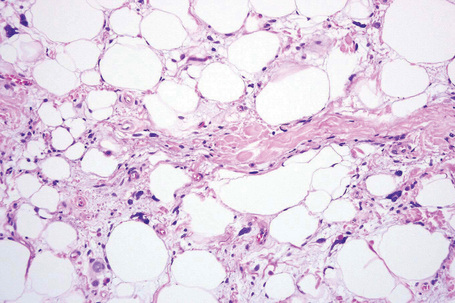
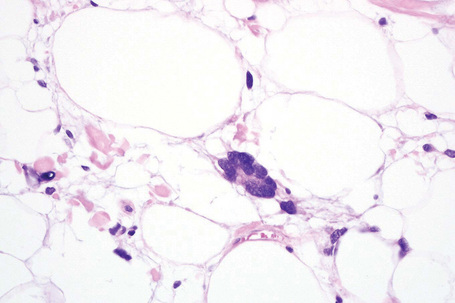
Chondroid lipoma
Clinical features
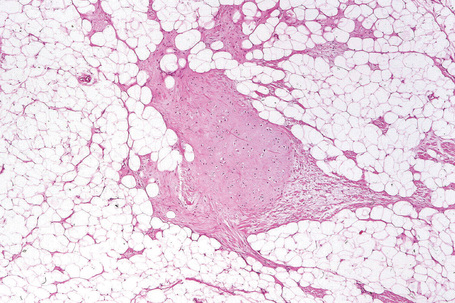
Chondroid lipoma is a distinctive tumor that presents predominantly in adult females, with a predilection for the proximal extremities and limb girdles.1 Tumors in children are very rare.2 Lesions are usually small and arise mainly in deeper soft tissues and (less commonly) in the subcutis. Rare examples present in the oral cavity, nasopharynx and pelvis.3–5 Clinical features are not distinctive. Tumors are benign and there is no tendency for recurrence after simple excision.
Pathogenesis and histological features
This tumor is characterized by t(11;16)(q13;p13) fusing C11orf95 and MLK2.6,7
The lesion is well circumscribed, lobular and often encapsulated. It consists of an admixture of mature adipocytes, uni- or multivacuolated lipoblasts and hibernoma-like cells with granular eosinophilic cytoplasm in a myxoid and chondroid matrix, which can show hyalinization (Figs 35.23, 35.24). Fibrosis and hemorrhage are often seen. The matrix is composed of chondroitin sulfate.8 Some tumor cells contain glycogen in the cytoplasm. Ossification is exceptional.9
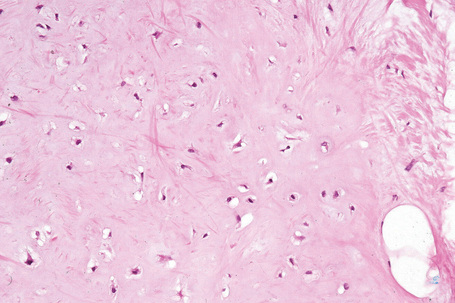
By immunohistochemistry, mature adipocytes are strongly positive for S-100 protein; lipoblasts are only weakly positive for this marker.8 Cytokeratin is very rarely focally positive.8
Electron microscopy confirms the presence of lipoblasts and mature fat cells. There is no evidence of true cartilaginous differentiation.10
Lipomatosis
Clinical features
Lipomatosis is extremely rare and may present in several forms, two of which affect the superficial subcutaneous tissue:1–4
A localized form of lipomatosis of the scalp has been reported as encephalocraniocutaneous lipomatosis and is associated with alopecia and ocular and cranial abnormalities.18,19
Congenital facial infiltrating lipomatosis refers to a disorder associated with hypertrophy of bones and soft tissues, macrodontia and premature dental eruption.20,21
Adiposis dolorosa
Clinical features
Adiposis dolorosa (Dercum’s disease) is a rare disease characterized by painful, circumscribed areas with increased fat in a plaque-like distribution.1–3 Usually, multiple body sites are involved but there is predilection for the buttocks, lower limbs and abdomen, particularly in juxta-articular areas. Localized disease is very rare and the breasts may be involved.4,5 The condition is much more common in females, especially after the menopause, and patients often have associated obesity and psychological problems.1–3 Occasional cases are inherited in an autosomal dominant manner.6,7 A case associated with the use of corticosteroids and a further patient with hypercholesterolemia and severe atherosclerosis have been documented.8,9 Pain occurs mainly as the result of palpation but may also occur spontaneously or as a result of movement.
Nevus lipomatosus superficialis (Hoffman and Zurhelle)
Clinical features
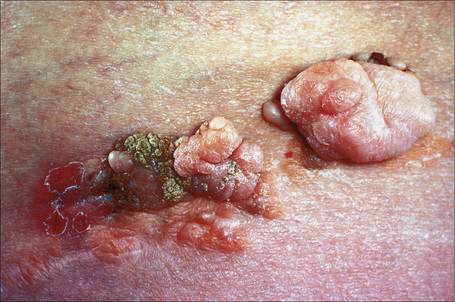
Nevus lipomatosus superficialis is an uncommon form of connective tissue nevus, manifest principally by the deposition of fatty tissue in the dermis.1–4 In its classical form, it is characterized by multiple papular, polypoid or plaque-like lesions, up to 2 cm in diameter, which almost always arise unilaterally on the posterior surfaces of the buttocks, upper thighs or lower back. More extensive and diffuse involvement may occur and patients present with prominent folds in what has been described as the Michelin tire appearance.5–7 Typically, the lesions present in early childhood or adolescence. Unusual associations include co-occurrence with lipedematous scalp, folliculosebaceous cystic hamartoma, dermoid cysts and angiokeratoma of Fordyce.8–11 A solitary form, usually seen in adults, shows a predilection for the same sites or occurs elsewhere and is more likely to represent a variant of fibroepithelial polyp or skin tag (Figs 35.25, 35.26). Such lesions have been described as pedunculated lipofibroma.12 In all types of nevus lipomatosus the sex incidence is equal. The papules or plaques, varying from skin-colored to yellow, are characteristically broad based and may show superficial comedone formation.
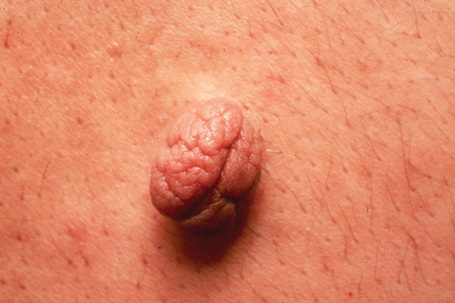
Fig. 35.25 Nevus lipomatosus superficialis: solitary lesions are often polypoid and have a soft consistency.
By courtesy of the Institute of Dermatology, London, UK.
Pathogenesis and histological features
The pathogenesis is unknown. In a single case, a 2p24 deletion has been described.13
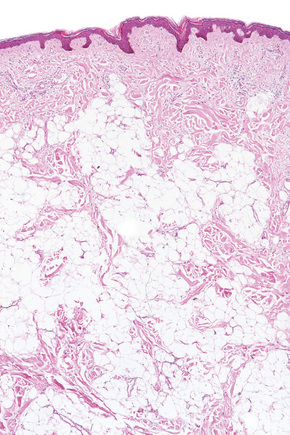
Although alterations are seen in all the connective tissue elements in the dermis, the predominant feature is deposition of lobules of mature fat in variable quantities in the superficial dermis (Fig. 35.27).3,14 These fatty lobules are located particularly around small blood vessels, the numbers of which are also increased. Areas of loose fibrous tissue, diminished elastic fibers and reduced numbers of epidermal appendages may also be a feature.
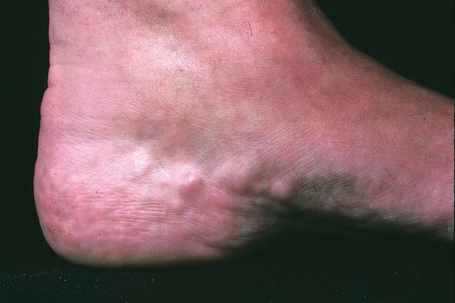
Piezogenic pedal papules
Clinical features
Piezogenic pedal papules characteristically present as multiple skin-colored papules on the heels (Fig. 35.28).1–3 They show predilection for the internal aspect of the heels and are usually asymptomatic although pain may be elicited when the patient is standing. Lesions become more noticeable when the patient stands up as a result of pressure. Piezogenic pedal papules are common in patients with Ehlers-Danlos syndrome.4 They have been described in athletes, including a marathon runner, and in association with Prader-Willi syndrome.5–7 Rare familial cases have been described.8
Lipomatosis of nerve
Clinical features
Lipomatosis of nerve (fibrolipomatous hamartoma of nerve, perineural fibrolipoma, perineural lipoma, intraneural lipoma) is a very rare hamartomatous condition usually occurring in children or young adults of either sex, around the wrists and hands, particularly along the distribution of the median nerve followed by the ulnar nerve and (less frequently) others including the brachial plexus and cranial nerves.1–5 Presentation at birth is sometimes seen. It can be associated with macrodactyly of the fingers innervated by the involved nerve in up to one-third of cases.1,6 Patients typically present with a slowly growing mass, which is either asymptomatic or associated with neurological symptoms including pain, paresthesia, loss of sensation or motor deficit. Carpal tunnel syndrome may develop when the median nerve is involved.7,8 Although the lesion is benign, treatment is difficult, as surgical excision often results in permanent neurological deficit.
Lipoblastoma
Clinical features
Lipoblastoma is the circumscribed subcutaneous counterpart of lipoma seen in infancy and childhood.1–6 Its diffuse form, lipoblastomatosis, is infiltrative and typically involves deeper structures, including muscle.
In either form, this condition most often presents in the first 9 years of life (exceptionally at birth, and 10% between the ages of 10 and 16), affects males rather more than females, and is typified by a slowly growing, usually subcutaneous mass with size ranging from 1 to 15 cm.1–7 Most tumors involve the trunk and extremities, followed by the head and neck.7,8 Presentation in the retroperitoneum, mediastinum and a number of internal organs including the kidney occurs rarely.9–14 Intrascrotal tumors and association with accessory scrota have also been documented.15–17 In the infiltrative type, local recurrence can occur following incomplete excision in up to 19% of cases.3 Familial cases may be seen.7 In about 17% of patients central nervous system anomalies are noted including macrocephaly, developmental delay, autism, Sturge-Weber syndrome and seizures.7 Unusual associations include a patient with glomuvenous malformations, epidermal nevus, temporal alopecia and heterochromia in addition to a lipoblastoma.18
A small series of adipose tumors closely resembling lipoblastoma and presenting in the vulva of young to middle-aged patients has been described as lipoblastoma-like tumor of the vulva.19
Pathogenesis and histological features
It has been shown that lipoblastoma has a consistent rearrangement of 8q11~q13, resulting in overexpression of the PLAG1 oncogene.20–29 Rarely chromosomal numerical abnormalities may be found.7,17,27,28
Lipoblastoma recapitulates developing fat and therefore contains varying proportions of mature adipocytes, lipoblasts and prelipoblasts arranged in a lobulated pattern and separated by loose fibrous connective tissue septa (Figs 35.30, 35.31). The stroma is often myxoid with numerous small capillaries giving a ‘crow’s feet’ appearance (Fig. 35.32) and may contain very primitive stellate or spindled tumor cells. Hibernoma-like tumors may be seen.30 Mitotic figures are uncommon; some cases show areas of extramedullary hematopoiesis.2
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


