Congenital Pyloric Stenosis and Duodenal Obstruction
Keith T. Oldham
Casey M. Calkins
In the years that have passed since the last edition of Mastery of Surgery, little has changed with regard to our knowledge of the etiology, pathophysiology, and diagnostic evaluation of patients with the conditions described in this chapter. Today, hypertrophic pyloric stenosis (HPS) is typically diagnosed with the aid of ultrasonography, and the treatment remains largely surgical, despite the occasional small series describing successful “less invasive” (atropine and intraluminal ballon dilation) means of treatment. Infants with pyloric stenosis represent a wonderful opportunity for the practicing surgeon to positively impact a patients’ life by undertaking a straightforward surgical intervention with excellent short and long-term outcomes. The laparoscopic approach to Ramstedt’s traditional open pyloromyotomy has been perfected, and many pediatric surgeons consider it a routine part of their therapeutic armamentarium. Regardless of the approach taken to pyloromyotomy, it remains one of the most gratifying operations that we perform as surgeons charged with the care of infants. Likewise, our understanding of the etiology and pathophysiology of duodenal obstruction and malrotation has been largely stagnant. The diagnostic approach to the infant with bilious vomiting remains unchanged—in that, malrotation and volvulus should be promptly addressed diagnostically, and volvulus treated as a surgical emergency. The specific intra-abdominal operative steps to surgically address the correction of duodenal obstruction and malrotation with or without midgut volvulus are largely no different than have been described in previous chapters addressing this same topic. However, the utilization of the minimally invasive
approach to both congenital duodenal obstruction and malrotation has increased since the last edition of this text, and we will address the technical aspects, as well as the possible benefits and pitfalls of these laparoscopic approaches herein.
approach to both congenital duodenal obstruction and malrotation has increased since the last edition of this text, and we will address the technical aspects, as well as the possible benefits and pitfalls of these laparoscopic approaches herein.
Congenital Pyloric Stenosis (Hypertrophic Pyloric Stenosis)
Introduction
HPS remains the most common surgical cause of nonbilious vomiting in infants. The incidence of pyloric stenosis ranges from 1 per 200 to 1 per 750 live births. Mode of inheritance is polygenic and modified by sex, with the incidence being four to six times higher in boys. Although it is believed to be more common in first-born males, birth order seems to be less influential than a smaller family size and higher socioeconomic status. The typical age of presentation occurs between 3 and 8 weeks, with a peak occurrence at 3 to 5 weeks of age. Rare cases of HPS diagnosed at birth or in utero have been reported. Only 4% of cases occur beyond the age of 3 months. Approximately 10% of cases are seen in previously premature infants. Premature infants with HPS typically present approximately 2 weeks later compared to term infants and the progression of the vomiting often evolves more slowly, frequently leading to a delay in diagnosis. In particular, preterm infants who receive intravenous prostaglandin therapy and transpyloric feedings appear to be more prone to pyloric stenosis. Patients typically present with postprandial, forceful, nonbilious “projectile” emesis, and the infant remains hungry and eager to feed after vomiting. Many infants are initially believed to have a food allergy or gastroesophageal reflux and the diagnosis may be delayed until the vomiting consistently follows every feeding and is forceful. The emesis may become bloody or appear as “coffee grounds” as a consequence of gastritis or esophagitis. Prolonged vomiting leads to dehydration, weight loss, gastritis with resultant hematemesis, and hypochloremic hypokalemic alkalosis. In modern practice, increased awareness among pediatricians and family practitioners along with the emergence of ultrasound as a highly accurate and noninvasive imaging study has resulted in earlier diagnosis, and 90% of affected infants are initially seen without serious dehydration or electrolyte disturbance.
HPS is the manifestation of progressive hypertrophy of the muscular wall of the gastric outlet. Though a single unifying etiology is unclear, several theories have been proposed including decreased nitric oxide synthase production in the enteric neurons within the pyloric musculature, a lack of interstitial cells of Cajal to allow relaxation of the pyloric channel, infectious and environmental agents, hypergastrinemia, increased synthesis of epidermal growth factor, and congenital redundancy of the pyloric mucosa. To date, none of the theories proposed offers a definitive answer to the question of the etiology of HPS. Whatever the mechanism for development of pyloric stenosis, it must take into account that the process of HPS generally occurs several weeks after birth, and the muscle hypertrophy is transient even without myotomy.
The occurrence of true idiopathic hypertrophic pyloric stenosis in adults is seldom encountered, but must be considered in the differential diagnosis of an adult with gastric outlet obstruction. Only 200 cases have been reported in the medical literature. Both pyloromyotomy and endoscopic dilation have been described as successful treatments. It is unclear if this entity represents a failure to recognize a “low grade” type of pyloric hypertrophy that has been present for life, or a new onset adult form of hypertrophic pyloric stenosis.
Clinical Presentation and Diagnosis
The diagnosis of pyloric stenosis can still be made by a careful history and physical examination. Observation of abdominal peristalsis with projectile nonbilious vomiting, and palpation of a mobile pyloric mass (the “olive”) is pathognomonic. This mass is palpable to an experienced examiner in up to 80% to 90% of cases, but may be difficult to appreciate in the face of a dilated stomach or irritable infant. Gastric decompression and a quiet baby (giving the infant a dextrose-coated pacifier) may aid in examination, yet few surgeons possess the skill and patience required to be considered “experienced examiners” when it comes to appreciating the olive on examination. Today, the diagnosis is often confirmed by ultrasonography, which is both specific and sensitive, with a 100% diagnostic accuracy in the hands of skilled radiologist or technician. A pyloric channel with a thickness >3 mm and a length >2 cm, are diagnostic hallmarks. The radiologist may also comment on the failure of fluid to pass through the pyloric canal, which is suggestive, but not specific for HPS. When the examination findings are equivocal, or additional tests are necessary to confirm one’s clinical suspicion, an upper gastrointestinal (UGI) contrast studies can be obtained. The classic UGI demonstrates a partial gastric outlet obstruction or “string-sign.” An adequate study requires some contrast in the duodenum to clearly establish the extent of the pylorus and differentiate pyloric stenosis from pylorospasm, which can also cause complete gastric outlet obstruction. An additional advantage of an UGI is its ability to evaluate other conditions, such as intestinal malrotation, duodenal stenosis, or gastroesophageal reflux disease, if the characteristic findings of pyloric stenosis are not present. In particular, if there is a history of bile-stained emesis, an upper GI contrast study should be performed expeditiously to evaluate malrotation of the intestines and midgut volvulus, a potentially life-threatening surgical emergency in infants. Once the diagnosis of HPS is made, the patient is NOT immediately taken to the operating room.
Preoperative Preparation
As many infants with pyloric stenosis present with dehydration, fluid resuscitation remains the single most important priority before the initiation of a therapeutic procedure. The initial evaluation of any infant with vomiting should focus on hydration status and serum electrolytes, including blood glucose, as severe dehydration can develop rapidly in infants and the consequences include cardiovascular collapse and hypoglycemic seizures with potential for permanent neurologic injury. Although patients with HPS are classically hypokalemic, it is always wise to keep potassium out of any replacement intravenous fluid in any patient with severe volume contraction until one is clear about the serum potassium. Therefore, when placing the IV, blood work should be obtained for sodium, potassium, chloride, and bicarbonate. Potassium should be added to the intravenous fluid once normo- or hypokalemia has been established. The classic metabolic derangement is that of hypokalemic, hypochloremic metabolic alkalosis. This can be profound in some patients, but is generally mild owing to the prompt recognition of the diagnosis amongst primary care providers. Correction of both alkalosis and hypokalemia is imperative before proceeding with surgery—which is nonemergent. Early, the loss of gastric secretions from vomiting leads to dehydration and aldosterone-stimulated potassium excretion in the urine, in an attempt to conserve sodium. As potassium depletion worsens, hydrogen ion is exchanged for sodium across the renal tubule, resulting in the “paradoxical aciduria” characteristic of patients with dehydration and metabolic alkalosis
from protracted vomiting. Mild dehydration and electrolyte disturbances can be corrected preoperatively over 12 to 24 hours or less with 0.45% normal saline with 5% dextrose and 20 mEq/L of potassium chloride. Rarely, severe disturbances may require initial correction with a 0.9% normal saline bolus (10 to 20 mL/kg) followed by an infusion of 0.9% normal saline in 5% dextrose at rates 25% to 50% above maintenance (100 mL/kg/d) and correction over a longer period. Infants should always have intravenous fluids with dextrose because of their limited reserves for gluconeogenesis and their vulnerability to hypoglycemia. Severe physiologic abnormalities should be corrected slowly to avoid seizures and other complications associated with rapid fluid and electrolyte shifts. Generally accepted parameters that should be met before operative correction of the obstruction include urine output >1 mL/kg/h, serum chloride >95 mEq/dL, serum sodium >135 mEq/dL, and serum bicarbonate <30 mEq/dL because of potential myocardial dysfunction and respiratory depression when it is elevated above this level. In addition, failure to correct the alkalosis before the operation frequently results in apnea on emergence from anesthesia. The patient is maintained NPO. We do NOT place a oro- or nasogastric tube, as continued removal of intragastric contents by suction only serve to further exacerbate the metabolic derangement.
from protracted vomiting. Mild dehydration and electrolyte disturbances can be corrected preoperatively over 12 to 24 hours or less with 0.45% normal saline with 5% dextrose and 20 mEq/L of potassium chloride. Rarely, severe disturbances may require initial correction with a 0.9% normal saline bolus (10 to 20 mL/kg) followed by an infusion of 0.9% normal saline in 5% dextrose at rates 25% to 50% above maintenance (100 mL/kg/d) and correction over a longer period. Infants should always have intravenous fluids with dextrose because of their limited reserves for gluconeogenesis and their vulnerability to hypoglycemia. Severe physiologic abnormalities should be corrected slowly to avoid seizures and other complications associated with rapid fluid and electrolyte shifts. Generally accepted parameters that should be met before operative correction of the obstruction include urine output >1 mL/kg/h, serum chloride >95 mEq/dL, serum sodium >135 mEq/dL, and serum bicarbonate <30 mEq/dL because of potential myocardial dysfunction and respiratory depression when it is elevated above this level. In addition, failure to correct the alkalosis before the operation frequently results in apnea on emergence from anesthesia. The patient is maintained NPO. We do NOT place a oro- or nasogastric tube, as continued removal of intragastric contents by suction only serve to further exacerbate the metabolic derangement.
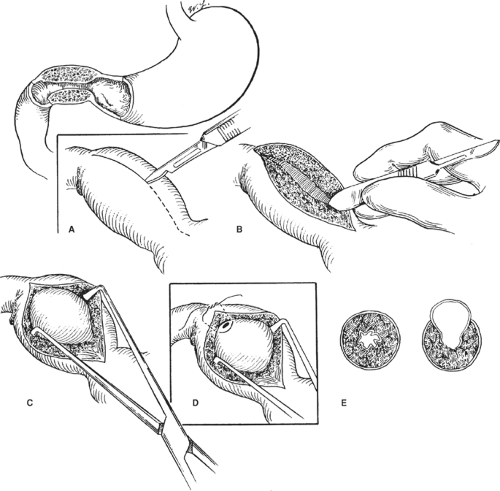 Fig. 1. Pyloromyotomy. A: The superficial serosal incision is begun on the duodenal end of the hypertrophied pylorus and carried onto the stomach. B: The scalpel handle divides the fibers. Note asymmetry of upper and lower muscle, indicating a complete myotomy at the duodenal end. C: The pyloric spreader further mobilizes the muscle. D: A perforation is closed with a horizontal mattress suture. E: The completed myotomy shown in cross section with a “bulging” pyloric mucosa. |
Operative Technique
Open
Classically, a transverse (2.5 to 3 cm) right upper quadrant muscle-splitting incision is used midway between the umbilicus and the xiphoid (Fig. 1). A supraumbilical skin incision, with a midline fascial extension can also be used with equivalent results. The liver edge is retracted superiorly, the greater curvature of the stomach is identified, and a point away from the pylorus is delivered into the wound. Gentle rightward
traction is placed on the greater curvature until there is adequate stomach to grasp with a moistened sponge, after which gentle leftward traction delivers the pylorus. The pylorus typically has a pale white appearance and a rubbery texture. The pyloric channel is controlled between the index finger and the thumb, and the junction between the pylorus and the duodenum is identified. The pyloric vein is a consistent anatomic marker just proximal to the pyloric–duodenal junction. The serosa is incised superficially on its anterosuperior aspect in an avascular plane beginning 1 to 2 mm proximal to the pyloric vein, or the obvious color change between the pale pylorus and the pink duodenum, and extended proximally onto the stomach beyond the proximal extent of the hypertrophied muscle. The myotomy is initially deepened to the level of the submucosa using the back end of a scalpel handle, starting in the thickest central portion and splitting the muscle fibers until the mucosa protrudes between the walls of the muscular defect. The pyloromyotomy is completed toward the stomach and duodenum using a Benson pyloric spreader (or a small right angle or hemostat) placed in the pyloromyotomy incision with the blades on the pyloric muscle, and the cut edges of the muscle are distracted in a carefully controlled manner. Care must be taken to avoid perforation of the underlying mucosa, particularly at the duodenal end where the duodenal mucosa prolapses over the shoulder of the hypertrophied pyloric muscle. Further disruption of the muscle occurs proximal and distal to the spreader, allowing further prolapse of the pyloric mucosa into the defect. Pyloromyotomy is considered complete when the superior and inferior borders of the pyloric channel move independently. The superior muscle edge is typically above and distal to the inferior muscle edge. Using this sign as a guide to completion of the myotomy avoids overvigorous distraction, which can lead to mucosal perforation.
traction is placed on the greater curvature until there is adequate stomach to grasp with a moistened sponge, after which gentle leftward traction delivers the pylorus. The pylorus typically has a pale white appearance and a rubbery texture. The pyloric channel is controlled between the index finger and the thumb, and the junction between the pylorus and the duodenum is identified. The pyloric vein is a consistent anatomic marker just proximal to the pyloric–duodenal junction. The serosa is incised superficially on its anterosuperior aspect in an avascular plane beginning 1 to 2 mm proximal to the pyloric vein, or the obvious color change between the pale pylorus and the pink duodenum, and extended proximally onto the stomach beyond the proximal extent of the hypertrophied muscle. The myotomy is initially deepened to the level of the submucosa using the back end of a scalpel handle, starting in the thickest central portion and splitting the muscle fibers until the mucosa protrudes between the walls of the muscular defect. The pyloromyotomy is completed toward the stomach and duodenum using a Benson pyloric spreader (or a small right angle or hemostat) placed in the pyloromyotomy incision with the blades on the pyloric muscle, and the cut edges of the muscle are distracted in a carefully controlled manner. Care must be taken to avoid perforation of the underlying mucosa, particularly at the duodenal end where the duodenal mucosa prolapses over the shoulder of the hypertrophied pyloric muscle. Further disruption of the muscle occurs proximal and distal to the spreader, allowing further prolapse of the pyloric mucosa into the defect. Pyloromyotomy is considered complete when the superior and inferior borders of the pyloric channel move independently. The superior muscle edge is typically above and distal to the inferior muscle edge. Using this sign as a guide to completion of the myotomy avoids overvigorous distraction, which can lead to mucosal perforation.
With the original incision having been placed in the most avascular portion of the pylorus, attempts at hemostasis of the cut edges are unnecessary. Oozing is a result of venous congestion associated with having the stomach out of the abdomen through a small incision and resolves spontaneously on return of the stomach to the abdominal cavity. A careful assessment of mucosal perforation is performed by gentle “milking” of the duodenum and antrum at the extents of the myotomy and inspection for any leaking air, bile, or gastric contents before returning the pylorus to the abdominal cavity. Unrecognized perforations are associated with significant morbidity, whereas the management of recognized perforations is generally straightforward. The wound is closed in layers with absorbable suture and the skin is closed with a subcuticular suture. Attention to detail is important during the wound closure as pyloromyotomy has a significantly higher than expected incidence of wound complications, including fascial dehiscence.
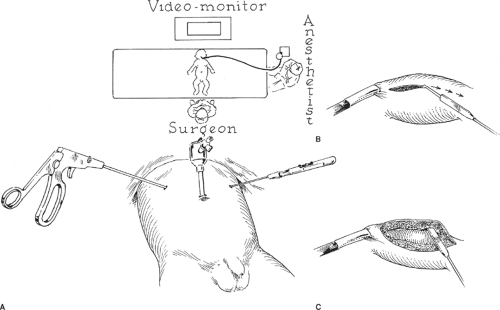 Fig. 2. Laparoscopic pyloromyotomy. A: Position of monitor, surgeon, patient, and trocars. B: The superficial incision is made using the sheathed arthroscopy knife with the sheath withdrawn. C: The myotomy is created by pulling with the arthroscopy knife blade sheathed. |
Laparoscopic
Laparoscopic pyloromyotomy has been demonstrated as a safe approach with results at least equivalent to the open surgical procedure and may result in decreased time to full feeding, less perioperative pain, and a superior long-term cosmetic result. After induction of general anesthesia, the infant is placed across the operating table (Fig. 2). The surgeon stands at the patient’s feet, while the camera
tower is placed at the patient’s head on the other side of the operating table. The abdomen is insufflated to 6 to 8 mm Hg through a Veress needle inserted at the umbilicus or through an umbilical trocar (5 or 3.5 mm) inserted by the open technique. We utilize an open technique, which is easy to accomplish in an infant. A 0- or 30-degree telescope (5 or 2.7 mm) is introduced through the umbilical port. Under direct vision, a stab incision is made in the right upper quadrant and a 3-mm grasper is introduced directly (without a trocar) to grasp the first portion of the duodenum and stabilize the pylorus. A sheathed arthroscopy knife is inserted through a second similar stab incision in the left upper quadrant. Some prefer to grasp the stomach for stabilization rather than the duodenum because it is less likely to be injured—in which case the instruments are inserted accordingly. The arthroscopy blade is extended, and the standard incision is made in the pylorus along an avascular plane. The blade is then retracted and the blunt sheath is used to widen the myotomy down to the mucosa. A laparoscopic spreader is utilized in a similar manner to the Benson spreader utilized in the open operation. Alternatively, the upper and lower portions of the muscle may be grasped with two graspers and slowly separated from one another with countertraction. The two edges are separated until the myotomy is deemed complete in the same manner as described for the open approach. The stomach is insufflated with 30 to 60 mL of air through an orogastric tube while occluding the proximal duodenum with a grasper to check for mucosal perforation. The instruments are removed and the wounds are closed. The laparoscopic approach continues to gain acceptance amongst pediatric surgeons. Two prospective, randomized controlled trials have demonstrated its equivalent safety and efficacy when compared to the open approach. However, there is a rather steep learning curve to the operation as it pertains to the completion of the myotomy, and should be undertaken by those with considerable pediatric laparoscopic experience or with specialized proctoring by an experienced surgeon.
tower is placed at the patient’s head on the other side of the operating table. The abdomen is insufflated to 6 to 8 mm Hg through a Veress needle inserted at the umbilicus or through an umbilical trocar (5 or 3.5 mm) inserted by the open technique. We utilize an open technique, which is easy to accomplish in an infant. A 0- or 30-degree telescope (5 or 2.7 mm) is introduced through the umbilical port. Under direct vision, a stab incision is made in the right upper quadrant and a 3-mm grasper is introduced directly (without a trocar) to grasp the first portion of the duodenum and stabilize the pylorus. A sheathed arthroscopy knife is inserted through a second similar stab incision in the left upper quadrant. Some prefer to grasp the stomach for stabilization rather than the duodenum because it is less likely to be injured—in which case the instruments are inserted accordingly. The arthroscopy blade is extended, and the standard incision is made in the pylorus along an avascular plane. The blade is then retracted and the blunt sheath is used to widen the myotomy down to the mucosa. A laparoscopic spreader is utilized in a similar manner to the Benson spreader utilized in the open operation. Alternatively, the upper and lower portions of the muscle may be grasped with two graspers and slowly separated from one another with countertraction. The two edges are separated until the myotomy is deemed complete in the same manner as described for the open approach. The stomach is insufflated with 30 to 60 mL of air through an orogastric tube while occluding the proximal duodenum with a grasper to check for mucosal perforation. The instruments are removed and the wounds are closed. The laparoscopic approach continues to gain acceptance amongst pediatric surgeons. Two prospective, randomized controlled trials have demonstrated its equivalent safety and efficacy when compared to the open approach. However, there is a rather steep learning curve to the operation as it pertains to the completion of the myotomy, and should be undertaken by those with considerable pediatric laparoscopic experience or with specialized proctoring by an experienced surgeon.
Operative Pitfalls and Complications
Complications after pyloromyotomy should be infrequent but include mucosal perforation, incomplete myotomy, wound infection, fascial dehiscence, and apnea. Vomiting, frequent in the early postoperative period, is thought to be a result of discoordination of gastric peristalsis or gastric atony related to gastric overdistension, and generally resolves within 48 to 72 hours. Abnormal or frequent vomiting, may suggest an incomplete myotomy or a missed perforation. A postoperative contrast study is performed if a leak is suspected, but is not helpful in evaluating the completeness of the myotomy because it takes several weeks for radiographic improvement in the setting of an adequate myotomy. Incomplete myotomy is rare, but typically occurs as a consequence of failure to carry the myotomy far enough onto the gastric antrum. Regardless of the approach taken to pyloromyotomy, one must be cognizant of the potential for significant morbidity and/or mortality when a mucosal perforation is missed. Mucosal perforation occurs commonly at the duodenal extent and can be repaired with fine absorbable sutures when recognized immediately. A patch of omentum is thereafter secured over the area akin to a Graham patch. Alternatively, the mucosal tear and myotomy can be closed, and a new pyloromyotomy performed after rotating the pylorus 180 degrees.
Postoperative Management and Outcome
The postoperative management of an uncomplicated pyloromyotomy is straightforward. Many different feeding regimens have been advocated with long-term equivalent success. Traditionally, there was a 12- to 24-hour delay after surgery to the initiation of feedings, and then small-volume, often dilute, feedings were begun and advanced as tolerated on a structured “graded” protocol. Recent reviews and prospective randomized studies have demonstrated early initiation and relatively rapid advancement of feedings to be equally well tolerated. We begin feedings 6 hours after operation (which is considered conservative in some circles), with low-volume balanced electrolyte or dextrose solution initially, and rapidly advance to full formula or milk ad libitum over a 12- to 24-hour period. Occasional emesis is common in the first 24 hours after pyloromyotomy, but this is typically self-limited and independent of the timetable or consistency of the feeding regimen and should not delay the progression of the feeding schedule in most cases. Persistent vomiting is most often a result of esophagitis or gastritis and is managed by stopping the feedings for several hours, and then restarting feedings at low volume. Significant postoperative vomiting coupled with physiologic deterioration should herald the possibility for a missed mucosal perforation, and an evaluation of the pyloric mucosal integrity undertaken immediately (either by UGI or operative exploration).
Outcome after pyloromyotomy is generally excellent. Most infants are discharged within 24 to 48 hours of surgery. Recent series of either open or laparoscopic pyloromyotomy have reported perforation rates of <2%, wound complications of 1%, and rare operative mortality. The pyloric muscle regresses to a normal thickness within 12 weeks following pyloromyotomy, and long-term prognosis is excellent. In a recent meta-analysis of the literature comparing open versus laparoscopic pyloromyotomy, time to full feedings and hospital length of stay was significantly lower in the laparoscopic group. In addition, the overall complication rate was lower in the laparoscopic group (although the mucosal perforation rate was equivalent between the two groups), without a significant difference in operative times.
Congenital Duodenal Obstruction
Introduction: Intrinsic Versus Extrinsic Obstruction
Congenital duodenal obstruction is a common problem in pediatric surgical practice with a reported incidence of 1 in 5,000 live births. An understanding of the various forms of congenital duodenal obstruction and frequently associated anomalies is essential to successful surgical outcomes. The obstruction may be complete (atresia) or partial (stenosis) and may be intrinsic to the duodenal wall due to a developmental abnormality, extrinsic due to compression of an otherwise normal duodenum, or a combination of both. Clinical recognition of duodenal obstruction generally occurs in the immediate neonatal period but occasionally, when the obstruction is incomplete, can be delayed until later childhood or even adulthood. Rotational anomalies are an important cause of duodenal obstruction in infants and children because of the attendant risk for volvulus and ischemic necrosis of the midgut. Malrotation can present at any age, including adulthood, but most commonly presents in the first month of life.
Intrinsic duodenal obstruction is the result of either complete or partial obstruction. Complete duodenal obstructions account for approximately 50% of all small intestinal atresias and are most commonly seen with continuity of the duodenal wall (type 1). Less commonly, the proximal atretic segment connects to the distal segment by a short fibrous cord along an intact mesentery (type 2), or there is complete separation of the proximal and distal duodenal segments associated with a
mesenteric defect (type 3). Partial or incomplete obstructions always have mural continuity and an intraluminal mucosal diaphragm or web, characterized by either a crescentic opening of variable size or a nearly complete membrane with a small central opening. Intrinsic obstructions can be challenging because there may be no external evidence of the precise location of obstruction other than a discrepancy in size between the proximal (dilated) and the distal (collapsed) segments. In addition, in a web with a small central patency, the endoluminal membrane may elongate due to peristalsis and high proximal intraluminal pressure, creating a situation in which the site of attachment of the membrane is several centimeters proximal to the apparent level of obstruction. This type of congenital duodenal obstruction has been referred to as a “windsock” deformity.
mesenteric defect (type 3). Partial or incomplete obstructions always have mural continuity and an intraluminal mucosal diaphragm or web, characterized by either a crescentic opening of variable size or a nearly complete membrane with a small central opening. Intrinsic obstructions can be challenging because there may be no external evidence of the precise location of obstruction other than a discrepancy in size between the proximal (dilated) and the distal (collapsed) segments. In addition, in a web with a small central patency, the endoluminal membrane may elongate due to peristalsis and high proximal intraluminal pressure, creating a situation in which the site of attachment of the membrane is several centimeters proximal to the apparent level of obstruction. This type of congenital duodenal obstruction has been referred to as a “windsock” deformity.
Extrinsic obstructions are the result of compression of an otherwise normal duodenum and, as with intrinsic obstructions, the obstruction can be partial or complete. There are many causes of congenital extrinsic obstruction of the duodenum including intestinal malrotation with Ladd’s bands or volvulus, preduodenal portal vein, gastroduodenal duplications, cysts or pseudocysts of the pancreas and biliary tree, and annular pancreas. Any of these anomalies may also be associated with an intrinsic duodenal obstruction.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


