CHAPTER 15 Congenital dyserythropoietic anemias
The congenital dyserythropoietic anemias (CDAs) are a heterogeneous group of rare inherited anemias, without additional cytopenias and with no tendency to neoplastic transformation.1,2 The cause of the peripheral anemia is a block in erythroid maturation, associated with a variable degree of erythroid dysplasia (dyserythropoiesis) in the bone marrow. The ineffectiveness of erythropoiesis results in a suboptimal reticulocyte response for the degree of anemia despite the presence of marked erythroid hyperplasia. Based largely on the nature of the dysplastic changes seen by light and electron microscopy, the CDAs have been divided into three major types, designated CDA types 1, 2 and 3 (Table 15.1), and other forms designated CDA groups IV–VII (see Table 15.2 below).3,4
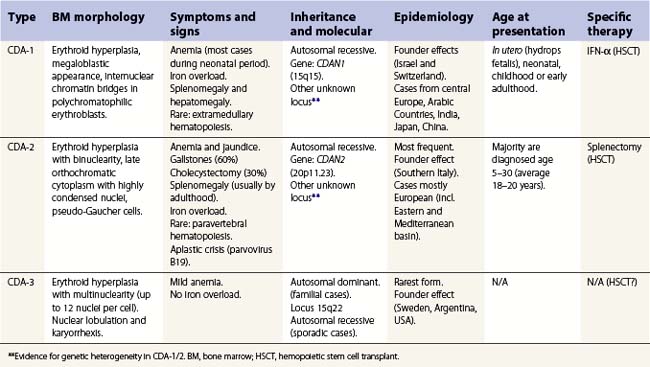
Table 15.2 Wickramasinghe’s CDA variants group classification
| For sake of clarity, groups of variants are indicated with Roman numerals, while major types are in Arabic numerals. To be considered a variant, at least three families must have been described with the features. First outlined by Sunitha Wickramasinghe (Wickramasinghe 2000).3 |
| Features | Inheritance | |
|---|---|---|
| Group CDA-IV | Severe transfusion-dependent normoblastic anemia with hypercellular marrow but absence of other features of CDA-1/2/3. If anemia is only moderate, then CDA-IVb. | Not clear |
| Group CDA-V | Mild normo/macrocytic anemia without medullary hyperplasia and erythroid dysplasia, but unconjugated hyperbilirubinemia. Has been reported in the literature as ‘primary shunt hyperbilirubinemia’ | Autosomal recessive/dominant |
| Group CDA-VI | Mild anemia with important macrocytosis (MCV 120–130 fl) and marked megaloblastoid hypercellular erythropoiesis. Orotic aciduria and thiamine-responsive anemia should be excluded. | Not clear |
| Group CDA-VII | As CDA-IV, but marked irregular nuclear shapes and with cytoplasmic globin-like inclusions (not reactive to α nor β antibodies). β thalassemia has to be excluded. | Not clear |
| Group CDA-VIII* | CDA with prominent post-splenectomy erythroblastemia (10–40 × 109 erythroblast/ml) but no resolution of a moderate anemia. Marrow showed irregular nuclei (clover-leaf forms) and karyorrhexis. | Not clear |
| Different variants | Reports of cases where classification and further delineation not possible. Many will possibly be classified within the spectrum of the major types when comprehensive molecular testing becomes available. |
* Previously identified as CDA with prominent post-splenectomy erythroblastemia.
Congenital dyserythropoietic anemia, type 1 (CDA-1)
CDA-1 has a variable presentation, as it can become apparent during the neonatal period or indeed at any time during the first five decades of life. It may also present in utero, with manifestations of fetal anemia requiring antenatal transfusions.5–7 However, patients usually have a mild to moderate anemia and may show jaundice, hepatosplenomegaly and cholelithiasis. A few are nevertheless severely anemic and transfusion-dependent. There is an increased tendency for some patients to develop marked iron overload despite a limited number of transfusions and lack of transfusion dependence (Fig. 15.1). Some cases have congenital abnormalities such as syndactyly, aberrations of the bones of the hands and feet (absence or hypoplasia of some phalanges, additional phalanges), dysplastic nails, short stature and abnormal pigmentation of areas of the skin (Fig. 15.2).
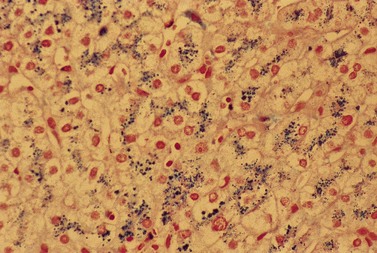
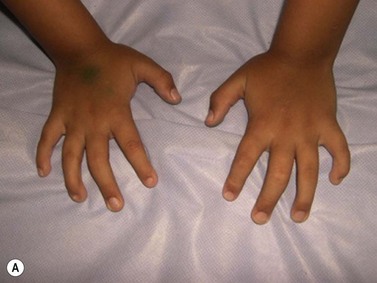
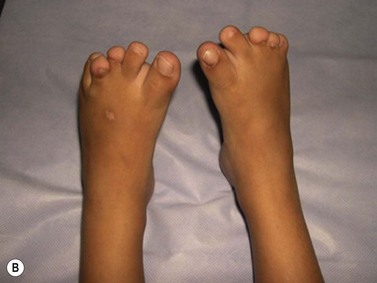
Fig. 15.2 (A, B) Dysmorphic features in the hands (A) and feet (B) of a child with CDA-1.
(Courtesy of Dr Aurora Feliu-Torres, Buenos Aires, Argentina.)
The disorder is inherited in an autosomal recessive manner. The disease gene was identified and localized to chromosome 15q15.1–15.3 in a highly inbred group of Israeli Bedouins.8 The CDAN1 gene has been highly conserved during evolution, has no known orthologues and the codanin-1 protein contains no recognizable motifs to provide clues as to its function. Over 20 mutations of CDAN1 have been described. The majority are missense mutations. Null mutations also occur, but to date no patients have been found who are either homozygous or compound heterzygous for these null mutations, suggesting such a combination is likely to be lethal. Incomplete sequencing may explain CDA-1 patients without CDAN1 mutations, but there is also likely to be genetic heterogeneity in this disorder.9
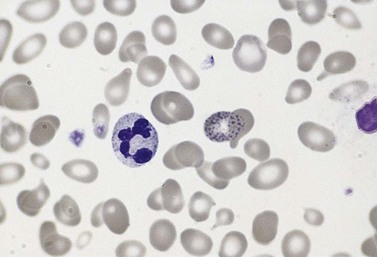
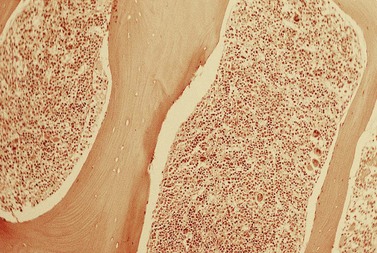
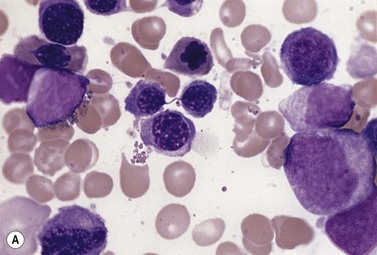
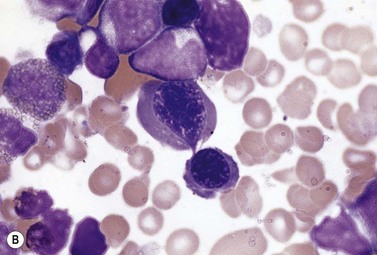
The average hemoglobin (Hb) is 9.0 g/dl (range 6.0–12.6 g/dl), and the mean cell volume (MCV) increased in 75% of cases. The peripheral blood film shows macrocytes and basophilic stippling (Fig. 15.3) and the reticulocyte count is normal or only minimally raised. There is megaloblastic erythroid hyperplasia in the bone marrow (Fig. 15.4). Some of the more mature basophilic erythroblasts and many of the early and late polychromatic erythroblasts show dyserythropoietic features. The characteristic morphological abnormality is internuclear bridging, where chromatin strands connect pairs of almost completely separated polychromatic erythroblasts (0.6–2.8 strands per 100 erythroblasts) (Fig.15.5A, B). There is also an increase in the percentage of binucleate polychromatic erythroblasts, which account for 3.5–7.0% of all erythroblasts. The latter may contain nuclei of different size and show partial fusion of the two nuclear masses; the two nuclei within the same cell may display different staining characteristics (Fig. 15.6). The most striking abnormality, seen on electron microscopy (EM), is the presence of nuclei with a spongy (or ‘Swiss-cheese’) appearance in a high proportion (up to 60%) of the mononucleate early and late polychromatic erythroblasts. These nuclei have multiple rounded electron-lucent areas within abnormally electron-dense heterochromatin (Fig. 15.7). Nuclei with a spongy appearance may also contain nuclear-membrane-lined cytoplasmic intrusions, sometimes with cytoplasmic organelles (Fig. 15.7B). An abnormally low percentage of mononucleate early polychromatic erythroblasts synthesize DNA; the non-DNA-synthesizing cells have DNA contents between 2c and 8c (1c = the haploid DNA content, i.e. the DNA content of a spermatozoon) and appear to be derived from the maturation of cells whose progress in the cell cycle has been arrested. These morphologically abnormal cells are destroyed in situ, resulting in ineffective erythropoiesis.10
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


