Male defects
Rectourethral fistula
Rectoperineal fistula
Rectovesical (bladder neck) fistula
Imperforate anus with no fistula
Rectal atresia/stenosis
Female defects
Rectovestibular fistula
Rectoperineal fistula
Cloacal malformation
Imperforate anus with no fistula
Rectal atresia/stenosis
Male Defects
Rectoperineal Fistula (Fig. 5.1)
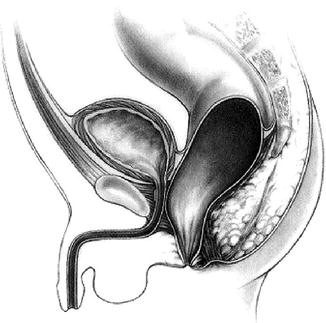
Fig. 5.1
Rectoperineal fistula (Figure 47.4, page 464 from Pediatric Surgery: Diagnosis & Management, Eds: Puri & Hollwarth, Springer 2009: ISBN 978-3-540-69559-2)
This is the lowest of the male defects. There is an opening of a fistula onto the perineum usually in the midline and almost always anterior to the sphincter mechanism. The opening may be as far anterior as the base of the scrotum and in some cases on the scrotum itself. Often the baby passes meconium through the fistula although this may take several hours following birth. The perineum is otherwise usually normal, and the likelihood of having an associated spinal or genitourinary abnormality is low. In the absence of an obvious opening or the appearance of meconium on the perineum, other clinical features that suggest the presence of this low type of malformation include a bucket-handle-type deformity or a subepithelial strand of white mucous. Selected cases may be repaired with a minimal anorectoplasty in the newborn period, thereby avoiding the need for colostomy.
Rectourethral Fistula (Fig. 5.2)
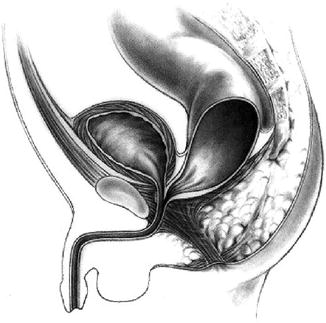
Fig. 5.2
Rectoprostatic fistula (Figure 47.2, page 464 from Pediatric Surgery: Diagnosis & Management, Eds: Puri & Hollwarth, Springer 2009: ISBN 978-3-540-69559-2)
The characteristic presentation of this type of defect is the absence of an anus with the appearance of meconium or meconium staining in the urine. There may be an anal dimple, well-developed buttocks and a midline groove with a bulbar urethral fistula but the chance of having such a ‘normal’ appearing perineum are lower with a fistula that opens into the prostatic urethra or higher. Internally, there is a common wall between the anterior wall of the fistula and the posterior wall of the urethra. Approximately one-third of cases with a bulbar urethral fistula have an associated genitourinary abnormality and this increases to two-thirds of cases with a prostatic fistula. All these infants require a diverting colostomy followed by a formal posterior sagittal anorectoplasty (PSARP) when the infant is a few months of age.
Rectovesical Fistula (Fig. 5.3)
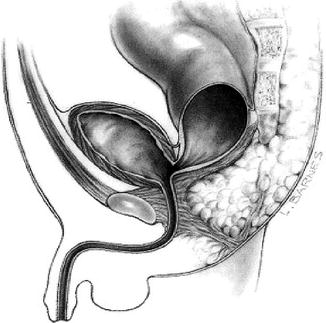
Fig. 5.3
Recto-bladder neck fistula (Figure 47.1 page 464, from Pediatric Surgery: Diagnosis & Management, Eds: Puri & Hollwarth, Springer 2009: ISBN 978-3-540-69559-2)
The fistula usually enters the bladder at the bladder neck and there may be meconium in the urine. The perineum is usually poorly developed with flat buttocks and underdeveloped muscles. There is a high incidence of associated urological and spinal anomalies. Internally the fistula enters the bladder most commonly in a T-shaped orientation without a long common wall. All infants with this high type of anomaly require a diverting colostomy followed by reconstruction when a few months older. The fistula is normally divided laparoscopically and the procedure completed from above rather than the usual approach from below.
Imperforate Anus Without Fistula
This is an uncommon anomaly (5 % of all male defects) and is commonly associated with Down’s syndrome. There is no meconium on the perineum nor in the urine, and the perineal muscle development is often reasonable. There is a low incidence of coexisting anomalies. These infants require colostomy formation in the newborn period followed by PSARP. Despite the absence of a fistula, there may be a long common wall between the rectal stump and the urethra which requires meticulous and complete dissection.
Rectal Atresia/Stenosis
These are the most rare of all anorectal anomalies and the least readily detected. Externally the anus appears entirely normal. Those with an atresia have a normal anal canal, an atresia 1–2 cm above the anal verge and a dilated blind pouch of proximal colon. In the case of rectal stenosis, there is a narrow, often fibrous passage between dilated colon and the anal canal. Both rectal atresia and rectal stenosis may be associated with a presacral mass as part of the Currarino triad. The perineum usually appears normal, but due to the dilated proximal colon, decompressive colostomy is recommended followed by later reconstruction. Because the anal canal is normal, functional outcome is generally good.
Female Defects
Rectovestibular Fistula (Fig. 5.4)
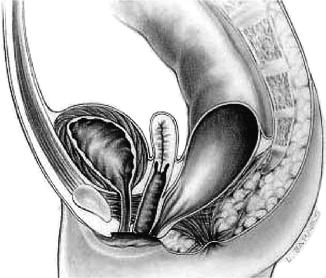
Fig. 5.4
Rectovestibular fistula (Figure 47.8, page 465 from Pediatric Surgery: Diagnosis & Management, Eds: Puri & Hollwarth, Springer 2009: ISBN 978-3-540-69559-2)
This, the most frequent defect seen in females, comprises a fistula opening into the posterior margin of the vaginal vestibule. The infant is usually able to pass some meconium through the fistula but not an adequate amount to allow decompression. The perineum is usually reasonably well developed, and the incidence of urogenital anomalies is approximately one-third, whilst sacral anomalies are rare. There is usually a long common wall between the fistula and the posterior wall of the vagina which requires meticulous and complete separation during anorectoplasty. The safest approach to an infant with a rectovestibular fistula is to form a colostomy. However, in a selected group of girls with this type of anomaly and an experienced surgeon, it may be possible to dilate the fistula adequately to allow decompression of the bowel prior to performing a primary PSARP without a covering stoma. This may be performed in the newborn period or, more easily, at a few months of age.
Rectoperineal Fistula
The female equivalent of this type of anomaly shares the features of its male counterpart. The fistula opens onto the perineum somewhere between the anal dimple and the vestibule and may often resemble a normal anus in appearance to the extent that some call this anomaly an ‘anterior ectopic anus’. Some cases do not require any surgical intervention so long as the calibre of the anus is adequate and the infant passes stool without undue difficulty. However, when constipation is problematic, a limited anorectoplasty without covering stoma may be performed.
Persistent Cloaca (Fig. 5.5)
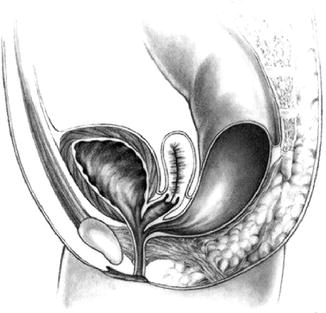
Fig. 5.5
Cloaca with common channel longer than 3 cm (Figure 47.6, page 465 from Pediatric Surgery: Diagnosis & Management, Eds: Puri & Hollwarth, Springer 2009: ISBN 978-3-540-69559-2)
Cloacal malformations are the most complex of all the congenital anorectal malformations. The vagina, urethra and rectum all open into a single common channel of variable length. This anomaly represents approximately 10 % of all anorectal malformations in females. There is a spectrum of severity determined primarily by the length of the common channel. This determines complexity of operative difficulty and functional outcome. With a short cloaca (defined as <3 cm in length), prognosis is generally good. However, in the presence of a channel >3 cm in length, there is a high incidence of vaginal and uterine anomalies, sacral anomalies and urinary tract obstruction. A common finding is vaginal obstruction resulting in hydrocolpos which may present as a palpable abdominal mass. The most significant morbidity associated with cloacas is that of obstructive uropathy with up to 30 % progressing to chronic renal failure.
All infants with cloacal malformations require a diverting colostomy and assessment of the urinary tract to ensure that drainage is adequate. On occasion a vesicostomy will be necessary. Hydrocolpos is treated with a vaginostomy. Formal repair occurs during the first year with a posterior sagittal anorectovaginourethroplasty (PSARVUP). For very high defects, an abdominal approach is required in addition to a perineal approach.
Other Female Defects
Imperforate anus without fistula and rectal atresia/stenosis are similar to their male counterparts and treated as such. Some classification systems include the female defect of rectovaginal fistula. In one large series of cases originally diagnosed as having a rectovaginal fistula and requiring reoperation, none were felt to actually have had such an anomaly. Two-thirds had features consistent with a diagnosis of persistent cloaca and the remaining third had a rectovestibular fistula.
Adequate and thorough neonatal assessment of all affected infants (including examination under anaesthetic if necessary) is essential to avoid erroneous surgical treatment with potentially adverse functional outcome. The key clinical features of a rectovaginal fistula are a clearly visible urethral orifice, a normally appearing vaginal orifice and meconium appearing from inside the vagina above the hymen. All such infants require initial colostomy. Detailed investigation and later reconstruction are performed when the baby is older.
Clinical Presentation and Diagnosis
The majority of anorectal anomalies present in the newborn period are detected at or soon after birth or during routine postnatal examination. The absence of an anus or an abnormal appearance of the perineum should draw attention to the underlying malformation. Surprisingly, however, it is not uncommon for the diagnosis to be missed and the infant to present with abdominal distension at a few days of age.
In addition to the abnormal external appearances, clinical features include absence of or minimal passage of meconium together with abdominal distension. In extreme circumstances, infants may present with intestinal perforation or urinary obstruction.
The diagnosis of an anorectal malformation is made on clinical grounds. In the majority of cases, adequate information can be achieved clinically to determine the likely anatomy and whether or not a colostomy is required. It is usual to wait 24 h before making any decision about surgical treatment. This allows time for meconium to appear at the perineum if this is possible. Some will require examination under anaesthesia in order to make a complete assessment.
In infants in whom the anatomy cannot be determined on clinical grounds (usually those in whom no meconium has been seen), some surgeons recommend taking a cross table lateral X-ray with the infant in the prone position (a so-called invertogram) once the child is 24 h of age. The position of the tip of the rectal stump on an invertogram may be seen as an air bubble. The proximity of this bubble to the perineal skin (which is marked using a radio-opaque marker) is used to guide subsequent management. If it is less than 1 cm, it is likely that there is an undiagnosed rectoperineal fistula that the defect is ‘low’ and a PSARP without colostomy may be possible. A bubble more than 1 cm from the perineal skin indicates a higher anomaly necessitating colostomy. If there is any doubt, the safest option is to perform a colostomy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


