At the time of its publication, section 503A of the U.S. Food and Drug Administration Modernization Act (FDAMA) served to define the limits of legitimate compounding.18 When section 503A of FDAMA was ruled unconstitutional in 2001, the delineation between compounding and manufacturing reverted to earlier regulations based on the Federal Food, Drug, and Cosmetics Act.19 Under those regulations, compounding is considered part of the practice of pharmacy and in most states, is governed by state law and regulation. Manufacturing is regulated by the federal government through the auspices of the Food and Drug Administration (FDA). In most cases, extemporaneously compounded preparations must be prepared pursuant to a prescriber’s prescription for a specific patient. Some states have specific regulations dealing with CSPs for office use. Some pharmacies whose primary purpose is preparing CSPs for hospitals and other facilities may be registered with the FDA as manufacturers and must adhere to federal good manufacturing practices. Some state boards of pharmacy permit one pharmacy to compound for another pharmacy under central fill regulations. Most pharmacies compound only pursuant to a prescriber’s prescription and follow state regulations regarding compounding.
On January 1, 2004, USP chapter 797, Pharmaceutical Compounding–Sterile Preparations,15 became official, replacing USP chapter 1206, Sterile Drug Products for Home Use.20 The change from a chapter numbered above 1000 to a chapter below 1000 marked a change from an advisory standard to an enforceable one. USP chapter 797 has since been revised.15 Some state regulations require full compliance with USP chapter 797, some have indirect references to the chapter, some do not mention the chapter, and some have additional regulations.21 The National Association of Boards of Pharmacy supports the incorporation of compounding regulations into state pharmacy practice legislation by including such wording in the association’s Model Rules and Model State Pharmacy Act.22 State boards of pharmacy should be consulted to determine the current status of sterile compounding regulations, as there are significant differences in regulation among states.
Accreditation Considerations
The Centers for Medicare and Medicaid Services (CMS) Hospital Conditions of Participation and Interpretive Guidelines, the Joint Commission, the American Osteopathic Association’s Healthcare Facilities Accreditation Program, and DNV Healthcare’s National Integrated Accreditation for Healthcare Organizations all include statements concerning safe practices for storage and preparation of sterile compounds.23–26 Clinics, long-term care facilities, home care organizations, rehabilitation facilities, and physician offices (all of which come under the purview of USP chapter 79715) may all be subject to specific additional governance of sterile compounding practices, depending on the agencies regulating or accrediting the facility. In addition, organizations preparing hazardous drugs27,28 should comply with National Institute for Occupational Safety and Health (NIOSH) recommendations to ensure that compounding personnel are operating in a safe environment.29,30
Other Compounding-Related Guidelines
ASHP provides several guidelines for safe compounding practices28,31,32 and a discussion guide on USP chapter 79717 and has recognized USP chapter 797 as a relevant practice standard in the ASHP Guidelines: Minimum Standard for Pharmacies in Hospitals.33 Other professional organizations also provide guidance on specific aspects of compounding. Standards for prescribing, preparation, administration, and monitoring of parenteral nutrition are available through the American Society for Parenteral and Enteral Nutrition.34,35 The Institute for Safe Medication Practices provides recommendations for preventing medication errors, including those involving CSPs.36,37 The Infusion Nurses Society offers standards, professional development, and resources for all aspects of infusion care.38 The Controlled Environment Testing Association (CETA) provides numerous CETA Application Guides (CAGs) for the proper use, cleaning, and certification of primary engineering controls (PECs) and buffer areas (generally referred to as “cleanrooms”).39–45 Guidelines for Hand Hygiene in Healthcare Settings,46 Guidelines for Prevention of Intravascular Catheter-Related Infections,47 Guidelines for Environmental Infection Control in Healthcare Facilities,48 and Protect Patients Against Preventable Harm from Improper Use of Single-dose/Single-use Vials,49 all from the Centers for Disease Control and Prevention (CDC), serve as the backbone for most infection prevention practices in the United States. Safe infusion, injection, and medication vial practices have been addressed by CMS50 and the Association for Professionals in Infection Control and Epidemiology,51 and the Association of peri-Operative Registered Nurses has recommended practices for medication safety in perioperative settings.52
Physical Facilities and Equipment
Design and Functionality Requirements
Facility requirements are intended to establish a safe environment for compounding CSPs. The International Organization for Standardization (ISO) air cleanliness classification of the compounding environment is a critical measure that is affected by facility design.
Primary Engineering Controls (PECs). A PEC is a device or room that provides an ISO Class 5 environment for compounding CSPs. PECs all rely on a special type of high-efficiency particle air (HEPA) filter that is ≥99.99% efficient in removing particles as small as 0.3 microns in size (the most penetrating particle size [MPPS], which refers to the largest-sized particle that may escape the filter, although particles of all sizes may be captured). The unidirectional (horizontal or vertical) HEPA-filtered air must provide sufficient velocity to sweep particles away from the direct compounding area and maintain unidirectional flow during preparation of CSPs. (More information about HEPA filtration and first-air concepts can be found in the ASHP publications Compounding Sterile Preparations,53 Basics of Aseptic Compounding Technique,54 Getting Started in Aseptic Compounding,55 and Compounding Sterile Preparations: ASHP Video Guide to USP <797>.56)
PEC devices include laminar airflow workbenches (LAFWs), biological safety cabinets (BSCs), compounding aseptic isolators (CAIs), and compounding aseptic containment isolators (CACIs) (Table 1). Properly designed, unidirectional airflow CAIs function in a similar manner as LAFWs, but the direct compounding area does not interact with room air because it is within a closed system, with the air sweeping particles away from the compounding site. Smoke tests of PECs assist a facility in verifying unidirectional airflow and lack of turbulence and reverse flows.
Primary Engineering Controls (PECs) | ||
PEC Device | Used to Prepare Non-Hazardous CSPs | Used to Prepare Hazardous CSPs |
Conventional | Laminar airflow workbench (LAFW) | Class II Biological safety cabinet (BSC) |
Isolators | Compounding aseptic isolator (CAI) | Compounding aseptic containment isolator (CACI) |
CAIs or CACIs located outside of an ISO Class 7 environment must be coupled with documentation from the manufacturer that the device will meet or exceed USP chapter 797 standards under these conditions and be dynamically tested on site to USP 797 and CETA requirements. If the CACI used for hazardous drug preparation is located outside the buffer area (see Architecture, below), it must be located in a segregated and dedicated area that maintains at least 0.01-inch water column negative pressure and maintains, at a minimum, 12 air changes per hour (ACPH).
Architecture. The sterile compounding area includes a well-lit buffer area and ante area (both are secondary engineering controls) and an area for storage of sterile products and supplies. A buffer area (or “cleanroom”) is defined as an area where a PEC is located and where activities such as preparation, compounding, and staging of CSPs occur. This area should provide adequate space for the PEC and may include a limited amount of shelving and/or carts for staging of compounding (not for storing stock). An ante area provides space for hand washing, garbing, and product decontamination; it also serves as a way to further segregate the buffer area from other, less-clean areas of the facility. Water sources, such as sinks or floor drains, are not permitted in the buffer area and should not be immediately adjacent to segregated compounding areas outside of a buffer area. A storage area outside the buffer and ante areas should provide adequate space for placement of sterile products and supplies.
The sterile compounding area (ante and buffer areas) may be constructed of either hard- or soft-walled enclosures, with the zones being delineated by open or closed architecture. Closed architecture is formed by walls and doors between the buffer and ante areas and is required for high-risk compounding (Table 2).
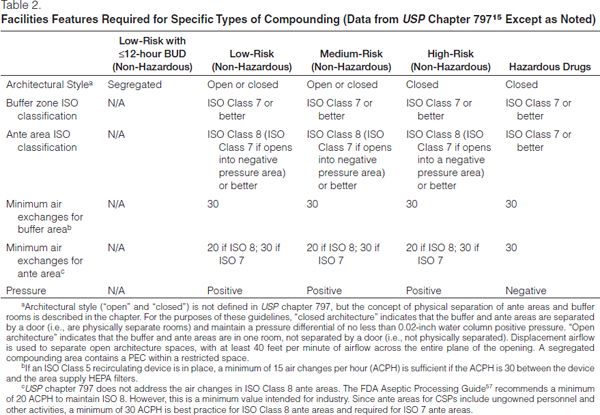
Open architecture has openings between the buffer and ante areas and relies on a defined airflow velocity to divide the two areas, which are marked by a line of demarcation; this type of facility may only be utilized for low- and medium-risk compounding. Demarcation lines should be indicated by colored tiles or other elements integrated into the flooring pattern but may be as simple as marking on the floor.
Facilities for preparation of radiopharmaceuticals have some different requirements. Refer to USP chapter 79715 and other relevant standards for specifics.
Facilities without USP chapter 797-compliant ante areas and buffer areas may prepare low-risk, non-hazardous CSPs in a PEC within a segregated compounding area. A segregated compounding area is an unclassified space (i.e., an area with no specific ISO classification) and does not include ante or buffer areas. It is required to be separated from activities that are not essential to the preparation of CSPs; not be located adjacent to food preparation sites, warehouses, or construction sites; and not have unsealed windows or doors that connect to the outdoors or high-traffic areas.15 This architecture type is most often seen in satellite pharmacies, small hospitals, procedural areas, or clinics. The beyond-use dating for sterile preparations compounded in a segregated compounding area cannot exceed 12 hours (see Expiration and Beyond-Use Dating).
Buffer Areas
Air Supply. A buffer area differs from an ordinary ventilated room by having the following:
- Increased air supply.
- HEPA filtration (the filtered air should be introduced at the ceiling, with returns mounted low on the walls; ceiling-mounted returns should not be used) including a terminal air filter (a filter at the end of the heating, ventilation, and air conditioning [HVAC] ducting).
- Room pressurization.
- A perforated plate or swirl supply air diffuser (if an air diffuser is necessary); high-induction supply air diffusers should not be used in buffer areas.
Structural components must be coupled with HEPA filtration and air exchanges in order to provide a complete buffer area environment and proper ISO classifications. Buffer areas must meet or exceed ISO Class 7 air cleanliness standards. Ante areas must at least meet ISO Class 8 standards; ante areas opening into a negative pressure preparation area must meet ISO Class 7 standards. The number of ACPH is based upon air/room pressure, velocity or air handler capacity, HEPA flow restriction, duct size, the amount of processing completed on a daily basis, and temperature. ACPH must occur at a minimum of 30 times per hour in buffer and ante areas, but may need to be increased in high-traffic/high-volume areas in order to maintain the room’s specified ISO classification (Table 2) under dynamic conditions. Facilities may incorporate the contribution of up to 15 air changes per hour from a LAFW in the total air changes per hours in a nonhazardous buffer area. By design, these devices filter room air as it passes through the HEPA filter.
Airflow within the room should be as steady as possible, having as few interruptions as possible. Within the PEC, it must be unidirectional,39 with as few interruptions in steady airflow as possible. PEC placement within the room should be well designed, with PECs placed where they are least affected by opened doors, HVAC systems, or personnel traffic. For non-hazardous preparations, positive pressure is required between rooms physically divided by walls or doors (closed architecture style) and should be maintained at a minimum of positive 0.02 inch water column. If a room does not have physical barriers (i.e., has an open architecture style) and relies on a line of demarcation, the displacement airflow concept requiring air velocity of 40 feet per minute (0.2 meter per second) from the buffer area across the entire plane of line of demarcation into the ante area is required. Open architecture is not permitted in areas used for high-risk preparations.
When designing buffer areas, facilities must consider workflow patterns, such as how personnel performing double-checks will affect air quality. If supervisory personnel are not located in the buffer area, movement in and out of the buffer area is likely to increase airflow interruption. Communication devices should be used to minimize traffic between areas, and cameras may be installed to supplement supervision of staff or check compounding accuracy, if permitted by state regulations.
Surfaces. Surfaces of any kind in the buffer area and ante area must be smooth, impervious, and easy to clean, with no cracks or crevices that could trap dust or contaminants. All materials used in the facilities must be non-shedding. Walls and ceilings must be made of either hard plastic or epoxy-painted gypsum board. If ceiling tiles are used, they must be coated with hard polymer and caulked both around the perimeter and around each tile. Ceiling lights must be smooth, mounted flush, and sealed. Floors should be made of wide, heavy-duty sheet vinyl, rubber, or epoxy that is coved around the corners and rolled up onto the walls. Paint must be an epoxy, acrylic, or other non-porous sealant type.
Work surfaces should preferably be stainless steel, but at a minimum are required to be non-porous and easily sanitized. Carts and shelves, ideally made of stainless steel wire, nonporous plastic, or rustproof metal, should be easy to move and clean, if necessary. Office equipment (e.g., computers and components [including washable keyboard and mouse], telephones, printers) placed in the buffer area must be easily cleanable and placed in such a manner that they have no material impact on the ISO air cleanliness classification of the area.
Renovations
To meet requirements for sterile compounding, many facilities choose to renovate existing space rather than construct new facilities. Whether designing a new area or retrofitting one, the specific types (e.g., hazardous or nonhazardous) and risk levels of CSPs that will be prepared in the area should guide the facility design and construction. A plan for how operations will continue without interruption should be devised prior to construction.
Power and Other Utility Interruptions
The facility’s emergency management plan should include steps to meet patient-care needs during time of utility interruptions, including the need for CSPs. In some cases, immediate-use procedures may be safely implemented to meet some needs. Methods to identify and safely meet interim compounding needs or address patient-care needs with noncompounded alternatives should be developed, put into standard operating procedures (SOPs), inserviced to staff, and tested as part of the organization’s emergency planning process.
Pharmacy Compounding Devices
Pharmacy compounding devices are utilized to increase efficiency while decreasing the potential for human error. Devices that do not create their own ISO Class 5 environment must be located within an ISO Class 5 PEC and adhere to applicable standards for accuracy and precision. All compounding devices must be monitored and validated for accuracy consistent with device manufacturer specifications.
Automated Compounding Devices (ACDs) are utilized to accurately combine multiple drugs and solutions into a single delivery container. These devices are most commonly used for parenteral nutrition preparation, but may be used for cardioplegia solutions, continuous renal replacement therapy, or other complex processes. ASHP Guidelines on the Safe Use of Automated Compounding Devices for the Preparation of Parenteral Nutrition Admixtures32 should be consulted for further details on utilizing ACDs. Accuracy and precision testing for ACDs is required by USP chapter 79715 and incorporate gravimetric, volumetric, and chemical analyses. These analyses, as determined by facility protocol, must be monitored and recorded on a daily basis, with evaluation for outliers occurring at least weekly.
Repeater pumps are devices used to pump a preset volume of fluid in a consistent and reproducible manner. They must be calibrated according to manufacturer specifications, which may depend on the volume and frequency of use.
Robotic systems automate the compounding and labeling of parenteral doses in syringes and bags using an enclosed chamber that must create an ISO Class 5 air cleanliness environment or better.
The proper use of ACDs, repeater pumps, robotic systems, and other compounding equipment used in the preparation of CSPs remains the responsibility of the pharmacist.
Cleaning and Disinfecting
Cleaning with a germicidal detergent and water will remove visible solids or soiling before disinfecting. Disinfecting removes microbial contamination. It is critical that an appropriate germicidal detergent and water be used to clean all surfaces of the buffer and ante areas in addition to all of the PECs. Great care must be exercised to avoid getting the HEPA filters wet during cleaning. Cleaning with a germicidal detergent will leave a residue that needs to be removed from work surfaces (e.g., counter and PEC surfaces). This residue is best removed by using sterile 70% isopropyl alcohol (IPA).
Appendix II of USP chapter 79715 provides information on types of products that can be used for cleaning and disinfecting the ante and buffer areas, including floors, walls, and ceilings. Choice of cleaning and disinfection products should be approved by the organization’s appropriate authority (e.g., the Infection Control Committee).
Policies and procedures must be developed to ensure consistent practices, including dilution of cleaning products. Table 3 describes the minimum frequency for cleaning surfaces used to compound low- and medium-risk CSPs in the sterile compounding area.
Minimum Frequency for Cleaning of Specific Sites (Reprinted with Permission from USP Chapter 79715) | |
Site | Minimum Frequency |
ISO Class 5 PEC | Beginning of each shift Before each batch Every 30 minutes when compounding After spills When surface contamination is known or suspected |
Counters and easily cleanable work surfaces | Daily |
Floors | Daily |
Walls | Monthly |
Ceilings | Monthly |
Storage shelving | Monthly |
Environmental Monitoring
Environmental monitoring and related documentation must be completed on a routine basis to ensure adequate environmental and personnel controls are in place to prevent contamination of CSPs. Ensuring a safe compounding environment requires viable and nonviable airborne particle testing, pressure differential or displacement airflow measurement, temperature monitoring, and surface disinfection sampling and assessment. Nonviable particles are particles that do not contain a living organism, such as particles shed from paper or dust. Viable particles are living organisms, such as bacteria or fungal spores, that require nonviable particles to travel. Monitoring of humidity,39,44 sound,39 and lighting39 may also be considered by facilities to enhance the environmental monitoring program.
Each element of the monitoring program must be included in a sampling plan with sample locations, methods of collection, sampling frequency, and other specifics depending on the type of monitoring being performed. The environmental monitoring sampling frequency must occur at a minimum as listed below, with possible additional times based on the type of testing:
- At the commissioning and certification of new facilities and equipment.
- Every six months during routine re-certification of equipment and facilities.
- After any facility or equipment maintenance, including construction or remodeling of adjacent departments or work on shared air handlers.
- At any point when problems are identified with products, preparations, or employee technique or if a CSP is suspected to be the source of a patient infection.
Records of data collected through the monitoring program must be maintained as part of the overall quality assurance program of the facility. The data should be reviewed by management personnel or their designees and by the facility’s Infection Control Committee to ensure that the findings of the reports are addressed. Table 4 provides an overview of environmental monitoring requirements.
Environmental Monitoring Requirements (Adapted from USP Chapter 79715) | ||
Parameter | Monitored By | Frequency |
Temperature | Compounding personnel or facilities management staff (if electronic monitoring is centralized) | Documented daily (at a minimum) |
Pressure differential or velocity across line of demarcation | Compounding personnel | Documented each shift (preferably), daily (at a minimum) |
Qualified certifier | At least every 6 months | |
Nonviable particles | Qualified certifier | At least every 6 months |
Surface sampling | Compounding or laboratory personnel | Periodically, as defined by compounding and infection control personnel, at least every 6 months or after significant changes in procedures or cleaning practices |
Electronic device sample of viable particles | Compounding personnel or qualified certifier | At least every 6 months |
Temperature Monitoring. Any controlled temperature area used for compounding sterile preparations or for storage of sterile products or CSPs must be monitored at least once daily and results documented in a log. The facilities should maintain a comfortable room temperature (20°C [68ºF] or cooler) for properly garbed compounding personnel. If facilities use continuous temperature recording devices, they must be monitored and documented once daily to ensure they are functioning properly. Controlled temperature ranges are listed in Table 5.
Controlled Temperatures (Data from USP General Notices and Requirements58) | ||
Storage Condition | Centigrade | Fahrenheit |
Room temperature | 20 to 25°C | 68° to 77°F |
Cold temperature (refrigerated) | 2 to 8°C | 36° to 46°F |
Freezer (frozen) | –25 to –10°C | –13° to 14°F |
Pressure Differential or Air Displacement. Since positive- and/or negative-pressure rooms are required for sterile compounding, the appropriate differential pressure or air displacement velocities must be maintained. If closed architecture is used, a pressure differential between general, ante, and buffer areas must be monitored. A facility with open architecture design must monitor the differential airflow across the opening between ante and buffer areas.
A pressure gauge or velocity meter must be in place to monitor airflow between relevant areas. Pressure between ISO Class 7 positive-pressure areas and the general area must be at least 5 Pa (0.02-inch water column). Negative pressure areas should have no less than 2.5 Pa (0.01-inch water column) negative pressure to adjacent positive pressure. A monitored pressure indicator must be installed to ensure proper pressurization. If differential airflow is used as a measure, the velocity must be at least 0.2 meter per second (40 feet per minute).
Results of pressure differential and/or velocity of air displacement must be reviewed and documented each shift (at least daily) or by continuous device with alarms.
Nonviable Airborne Particle Testing Program. Determination of the ISO classification of an area or device is dependent on nonviable particle testing (“certification”), which must be completed by qualified personnel complying with the Certification Guide for Sterile Compounding Facilities (CAG-003–2006).39 PECs such as LAFWs, BSCs, CAIs, and CACIs must be certified every 6 months and whenever the device is relocated or serviced. Both primary (LAFWs, BSCs, CAIs, and CACIs) and secondary engineering controls (buffer areas and ante areas) must be checked for total particle counts every 6 months according to the manufacturer’s specifications or CETA recommendation and when a device or room is relocated or altered. Thresholds for each ISO class are presented in Table 6.

Viable Airborne Particle Testing Program. Classified space (PECs and buffer and ante areas) must undergo routine viable particle testing. The testing plan should include the required sample locations, method of collection, frequency, the volume of air to be tested, and the time of day testing will occur. Testing must occur every 6 months in all compounding areas (PECs, buffer areas, ante areas, and areas adjacent to segregated compounding areas) as part of the overall compounding recertification process. The method of testing must be impaction via an electronic air sampling device, as settling plates alone are not considered an acceptable method.
Sampling plans should be detailed and include all high-traffic locations within the compounding area and any sites prone to contamination. Turbulence caused by airflow disruption, such as within an ISO Class 5 LAFW or doorways, should be included in the testing plan, along with areas where garbing, cleaning, labeling, and staging occur. In segregated compounding areas, sampling should include locations within the ISO Class 5 PEC and other areas in close proximity to the PEC.
Viable particle testing must be performed using a general microbiological growth medium, such as sterile nutrient agar. In facilities that compound high-risk preparations, testing must also be done with a medium that supports fungal growth, such as malt extract. The growth medium should be incubated (outside of the sterile preparation area) according to the manufacturer’s recommendations.
Sample data must be reviewed as a means of evaluating control of the compounding environment. Results above recommended action levels (see Table 7) should prompt reevaluation of work practices, cleaning procedures, and HEPA filtration. Any microbial growth that results from viable environment sampling must be identified to the genus level by microbiology personnel. If any highly pathogenic organisms (e.g., gram-negative rods or yeasts) are identified, infection control specialists should immediately be consulted to assist in formulating a response to the situation.
Viable Environmental Monitoring Recommended Action Levels for Microbial Contamination (Adapted from USP Chapter 79715) | |
ISO Classification | Recommended Action Levels for Microbial Contamination (CFUs/m3)a |
5 | 1b |
7 | 10 |
8 or above | 100 |
aCFUs/m3, colony-forming units per cubic meter. bSamples from ISO Class 5 environments should normally yield no microbiological contaminants. | |
Surface Disinfection Sampling and Assessment. Touch contamination originating from contaminated work surfaces must be minimized and prevented if possible. Surface sampling provides facilities with a snapshot of the effectiveness of their disinfection procedures (including technique and cleaning products) and must be part of the overall quality assurance plan. Using a sterile nutrient agar contact plate for flat surfaces or swabs for equipment and other non-flat surfaces, sampling must be performed in all ISO classified areas on a periodic basis, at a minimum, every 6 months or when significant procedural or cleaning changes are implemented. A specific plan detailing the location of each sample must be devised so that the same locations are repeated with each testing session. Contact plates require pressing a plate directly to the surface being tested, while swabbing requires swabbing an area, submersing the swab in the correct amount of diluent, and then swabbing onto or into a sterile nutrient agar surface. Agar plates will leave a residue on contact surfaces that must be cleaned with sterile water and disinfected with sterile 70% IPA.
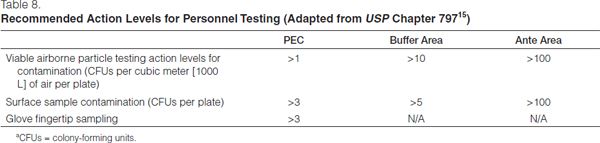
Results must be reported in colony-forming units (CFUs) per plate. Reevaluation of work practices and cleaning procedures should occur if the CFU count exceeds the suggested action levels (Table 8). Investigation into the source of contamination should be undertaken, the sources eliminated, and the area cleaned and re-sampled.
Environmental monitoring and quality assurance programs and documentation may be completed by a limited number of personnel in any given facility, but the actions of all compounding personnel may affect these two critical elements of compliance. All compounding personnel should be familiar with all facility policies and procedures specific to CSPs, even if the procedures are not typically their responsibility.
Expiration and Beyond-Use Dating
A manufacturer’s expiration date is the date assigned pursuant to manufacturer testing. The drug product is guaranteed by the manufacturer to be safe and effective up to the listed date when products are stored as described in the product labeling.
A beyond-use date (BUD) is the date or time after which administration of a CSP shall not be initiated. As described in previous ASHP guidelines14 and in USP chapter 797,15 the BUD is determined from the date or time the preparation is compounded, its chemical stability, and the sterility limits described later in these guidelines. Both the stability of the components and the sterility limits described above must be taken into consideration when determining BUDs, and the BUD must be the shorter of the sterility dating or chemical stability dating. Information regarding stability dating procedures and defaults can be found in USP chapter 795, Pharmaceutical Compounding—Non-Sterile Preparations59 and other published literature sources.60,61
Processes such as thin-layer chromatography (TLC) and high-performance liquid chromatographic (HPLC) assays are the most reliable means of determining the stability of a product and should be used in place of theoretical predictions of stability when published literature is not available. The use of commercial reference laboratories that offer qualitative and quantitative testing may serve as a key resource for end-product testing.
Risk Level Classification
In these guidelines, as in previous ASHP guidelines14 and USP chapter 797,15 CSPs are stratified by potential risk of microbial contamination into three primary categories: low-, medium-, and high-risk CSPs, with an additional category for CSPs intended for immediate use15
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


