Figure 34-1. The signal hypothesis of T-cell activation and molecular targets of modern clinical immunosuppression. T-cell activation requires three signals to initiate an effector response: antigen presentation of nonself MHC, costimulatory signal, and regulation of cytokine signaling at the cell surface. These three signals are thus targets for therapeutic drugs used in modern clinical immunosuppression in both induction and maintenance phases, as well as in the treatment of rejection. The figure demonstrates the site of action of these agents.
Memory T-cell subsets react in a more rapid manner to the graft and incite an accelerated rejection response. Seminal studies investigating memory responses to allografts proved that antigen-experienced memory T-cell subsets can cause graft destruction in the absence of secondary lymphoid tissue.9 Clinically, these “memory” T cells are generated throughout the lifespan of a potential recipient due to various infections that the patient may have experienced. Memory T-cell subsets that have been generated by these infections may then react against an organ at the time of transplant if the antigenic response from the organ mimics that of the previous infectious episodes. This concept of “heterologous immunity” may account for early rejection responses in patients who have not been classically sensitized by foreign human antigen by transfusion, pregnancy, or previous transplant.10 Studies have also suggested that certain induction therapies may eradicate naïve host T cells yet spare memory subsets which may also contribute to potential early rejection responses and express resistance to tolerance induction.11–13
Immunosuppression Induction in Solid Organ Transplantation
Human immunity depends on innate and adaptive responses to foreign antigen. The innate response is rapid and nonspecific when foreign antigen is encountered; the adaptive response develops over time and harbors the ability to recall pathogens upon each successive insult. Therefore, adaptive immunity becomes more efficient with each antigenic interaction. The innate immune response usually sets the scene for the adaptive response but it is the antigen specificity of the adaptive response that allows for activation and proliferation of effector cells to rid the system of the pathogenic insult. Current therapies in clinical immunosuppression target this global activation and proliferation of T cells.
3 Induction immunosuppression is used intraoperatively and/or perioperatively as a prophylactic treatment to attenuate acute rejection responses in the first few months after transplant when the risk is highest.14 Various induction strategies have been developed in order to spare the use of toxic calcineurin inhibitors (CNIs) and steroids.
CELL SURFACE RECEPTOR BLOCKADE
4 Each of the three signals in T-cell activation is uniquely important and each depends on the other for cellular proliferation and effector cell differentiation (Fig. 34-1).15,16 If one signal acts in isolation without the others, anergic or apoptotic signal ensues.17,18 With this knowledge, various pharmacotherapies have been developed to block signal 2, or costimulation, in order to inhibit the clonal proliferation of the effector T cell. Several pathways are involved in the make-up of the second signal in lymphocyte activation. Costimulation pathways have been targeted by various types of biologics including monoclonal antibodies as well as recombinant fusion proteins.
A specific APC-T–cell interaction is crucial. The engagement of APC-derived CD80/86 (B7 molecules) with CD28 on T cells (members of the Ig superfamily) is integral to positive costimulation.5 Pharmacotherapies targeting other costimulatory pathways are in various phases of development and include monoclonal antibodies to CD40 as well as CD2. Although costimulation blockade has been successfully utilized for the induction of tolerance in rodent models, this has not been realized in humans. Costimulation blockade has proven to be beneficial when used as maintenance immunosuppression.19
The initiation of signal 3 begins with the engagement of IL-2 on CD25 located on the surface of the T cell. IL-2 receptor blockade has become standard of therapy in the perioperative period (day 0 and day 4) to allow for early allograft acceptance and reduced usage of nephrotoxic CNIs. Early clinical trials proved that induction therapy with IL-2 receptor blockade significantly reduced acute rejection rates in renal transplant recipients when compared to placebo groups and in combination with standard CNIs and steroids.20
B7/CD28 CTLA4 Blockade
The expression of CD80 (B7.1) and CD86 (B7.2) on the surface of APCs provides T cells with either activation signals, if engaged to CD28, or inhibitory signals, if ligating cytotoxic T-lymphocyte antigen 4 (CTLA4). CD80 and CD86 interact preferentially with CTLA4 (CD152) when compared to CD28.18,21 CTLA4 is also up-regulated following the activation process and its ligation results in inhibition of activation thereby controlling clonal expansion.22,23 Additionally, CTLA4 is constitutively expressed on regulatory T cells.24 Blockade of each B7 molecule as monotherapy has not led to significant allograft survival in nonhuman primate models. In contrast, blockade of both B7 molecules together did lead to prolonged graft survival but was not able to induce tolerance as rejection occurred once therapy was stopped.25–27 An immunoglobulin fusion protein of CTLA4 (abatacept), has been shown to inhibit graft rejection in xenogeneic and allogeneic systems.28,29 These promising early results have prompted the development of CTLA4-Ig variants.
LEA29Y (belatacept) is a second-generation molecule, derived as a result of mutating two amino acids of CTLA4-Ig. Belatacept possesses a twofold greater ligation capacity for both CD80 and CD86. The increase in avidity allows for a more complete blockade of the B7 molecules and a 10-fold greater suppression in in vitro assays.30 Belatacept has shown improved renal function, reduction in chronic allograft nephropathy, decreased calcineurin-related toxicity, and no thromboembolic complications in phase II trials.31 Phase III trials have evaluated the efficacy and renal protection of belatacept in standard (BENEFIT) and expanded criteria donors (BENEFIT-Ext). There was an increased incidence of posttransplant lymphoproliferative disease in Epstein–Barr virus (EBV)-seronegative patients and acute rejection at 1 year in the belatacept treatment groups compared to CNIs. Additional results revealed an improved glomerular filtration rate (GFR) and lipid profile over a 5-year period.32–35
IL-2 Receptor Blockade
Basiliximab is a chimeric mouse/human IgG1 monoclonal antibody to the alpha chain of CD25 (IL-2 receptor) which has proven to reduce acute rejection rates in adult and pediatric renal and pancreas transplant recipients. In clinical trials with 6-month follow-up, basiliximab had a significant impact on acute rejection rates with no increase in infectious complications.36 Often used in kidney transplant patients as induction therapy, and in liver transplantation in order to spare the nephrotoxic effects of tacrolimus, long-term follow-up remains to be provided.37–39 In a randomized, prospective, placebo-controlled trial of patients investigating immunoprophylaxis in liver transplantation, investigators identified a lower acute rejection rate in HCV-negative patients at 6-month follow-up in patients treated with basiliximab in combination with standard CNI therapy.40
LEUKOCYTE DEPLETION
In order to achieve tolerance to an organ transplant, it may be necessary to maximize the potential of engraftment by depleting the alloreactive effector T cells (and B cells in some therapeutic regimens) which mediate a rejection response. Many investigators have identified the value of monoclonal or polyclonal antibodies designed to bind to peripheral alloreactive lymphocytes. The short-term depletion of leukocytes using antithymocyte globulin, anti-CD3, anti-CD2, anti-CD4, and anti-CD8 has proven successful in terms of long-term graft survival in animal models; these results are further enhanced with thymectomy prior to transplantation in order to prevent posttransplantation peripheral effector cell repopulation.41,42 In nonhuman primate models, anti-CD3 conjugated to diphtheria immunotoxin administered alone prior to transplantation or in conjunction with sirolimus (rapamycin) or deoxyspergualin, inhibitor monocytes and macrophages, produced tolerance via T-cell depletion.43–45 Clinical trials in kidney transplant recipients using anti-CD52 antibody (alemtuzemab)–mediated leukocyte depletion alone and in combination with deoxyspergualin were not able to induce tolerance.46,47 In subsequent trials, alemtuzumab therapy allowed for the use of low-dose immunosuppression in steroid-free regimens in order to control effector responses; alemtuzumab is now a commonly used and inexpensive induction agent.48–52 Memory T cells are the most resistant to depletion with alemtuzumab therapy.13,53
Polyclonal leukocyte-depleting antibodies, such as thymoglobulin (rabbit antithymocyte globulin [rATG]), are useful as induction agents, and are mainstays of induction therapy in renal transplant recipients.54 Upon binding various T-cell epitopes, rATG lyses leukocytes using complement-dependent pathways and opsonization leading to mononuclear cell phagocytosis of T cells.55 rATG also binds to surface receptors on dendritic cells, which impair antigen presentation.55 Polyclonal antithymocyte globulin is wrought with serious side effects including a predisposition to posttransplant lymphoproliferative disorder and the cytokine release syndrome.56,57
Depleting the recipient of leukocytes creates a window of opportunity for an allograft to “settle in” without attack by effector immunocytes. The recipient’s transient immunodeficiency is dictated by the strength and duration of depletion. T-cell depletion strategies have paved the way for future studies involving administration of donor antigen along with leukocyte depletion in an attempt to achieve chimeric states.58
CLINICAL THERAPEUTICS FOR MAINTENANCE IMMUNOSUPPRESSION IN SOLID ORGAN TRANSPLANTATION
Calcineurin Inhibitors
5 One of the greatest medical discoveries of 20th century was the identification of the CNIs with their effects on T-cell signaling, and their alterations in cell-mediated immunity. This class of agents has potent biologic activity and minimal myelosuppressive effects, which were a significant limitation in early clinical transplantation. Cyclosporine A was the first of these drugs identified, initially found in a soil sample from the fungus Tolypocladium inflatum. Cyclosporine is a cyclical fungal peptide composed of 11 amino acids, with significant immunosuppressive effects.59,60 Subsequently, the clinical efficacy of cyclosporine A was demonstrated in human kidney transplantation, opening the gates to a new era for clinical organ transplantation. Cyclosporine remained the mainstay of clinical immunosuppression for several years, and was used across organ types by thousands of patients. In 1984, FK506 (tacrolimus) was identified as another CNI with a similar mechanism of action. FK506 is a macrolide immunosuppressant that was approved by the U.S. Food and Drug Administration for use in liver transplantation.61–63 Tacrolimus has become the mainstay of maintenance immunosuppression in solid organ transplant programs due to a gentler side effect profile and potency of immunosuppression, and is used in virtually all solid organ types.
CNIs, as a class, disrupt regulatory T-cell signaling and lead to a down-regulation of IL-2 production, which eventually decreases the activity of cytotoxic T cells. Antigen presentation at the TCR leads to an increase in cytoplasmic calcium levels through a protein kinase and G-protein reaction within regulatory T cells. This calcium binds to calmodulin, a regulatory subunit of the phosphatase calcineurin, prevalent in immunocompetent lymphocytes and nerve cells. Calcineurin is responsible for cleaving the phosphate bound to NFAT (nuclear factor of activated T-cells) which in its activated form traverses the T-cell nuclear membrane and promotes transcription of IL-2. Inhibition of calcineurin occurs through the activation of immunophilins, such as cyclophilin and FK-binding protein, which are endogenous cytosolic peptides. The cyclosporine–cyclophilin complex and its counterpart, the FK506–FKBP12 complex, prevent dephosphorylation of NFAT, which in turn leads to reduced IL-2 production and diminished T-cell signal transduction to effector lymphocytes.
Side Effects
CNIs have a variety of side effects which present challenges in the clinical management of transplant recipients. One of the issues relates to drug interactions for patients on tacrolimus or cyclosporine. Common interactions that may boost levels of CNIs occur in patients requiring antifungal therapy, particularly in the azole class, as well as other drugs metabolized by hepatic/intestinal cytochrome P450 3A4 enzyme. As implied by the name, calcineurin is also present in nerve tissue as well as lymphocytes. Drug activity in nerve tissue leads to several clinical effects including tremors, seizures, and posterior reversible encephalopathy syndrome.64 Additional side effects include hyperkalemia, hypercholesterolemia, and gastrointestinal dysfunction including diarrhea. Cyclosporine has also been associated with gingival hyperplasia that is seen less frequently with tacrolimus. The most significant long-term effects include chronic nephrotoxicity that may precipitate end-stage renal disease, as well as new-onset diabetes mellitus after transplantation. Consultation with a transplant pharmacist is integral to the successful management of the posttransplant patient to prevent toxicity and achieve therapeutic immunosuppression goals.
mTOR Inhibitors
The first discovered mTOR inhibitor was sirolimus. Sirolimus, or rapamycin, is a bacterial macrolide derived from Streptomyces hygroscopicus, found initially in a soil sample from Easter Island (Rapa Nui). Like CNIs, its mechanism of action leads to decreased IL-2 proliferation. Sirolimus binds to FK-binding protein 12, as does tacrolimus. The site of action of the sirolimus–FKBP12 complex is known as the mammalian target of rapamycin, or mTOR. Despite having structural similarity to tacrolimus, sirolimus has limited activity on calcineurin when bound to FKBP12. The complex disrupts signal transduction of the IL-2 receptor and decreases nuclear transcription of molecules active in lymphocyte homing.65 Sirolimus also has significant antiproliferative properties, and inhibits cell cycle activity in lymphocytes and fibroblasts.66–68
Sirolimus has been adopted in a variety of clinical protocols. Sirolimus is currently used primarily in place of CNIs, most commonly in patients who are weeks to months out from the transplant operation.69 Sirolimus has less nephrotoxicity than CNIs, but has been associated with a higher risk of acute rejection in some studies. In liver transplantation, conversion from tacrolimus to sirolimus in patients with reduced kidney function was associated with more acute rejection episodes but did not have a tangible benefit in terms of kidney function in a randomized control trial.70 Some have advocated for lower-dose CNIs as maintenance therapy in patients with impaired renal function compared to sirolimus monotherapy,71 and studies have demonstrated the potential of low-dose CNIs in conjunction with sirolimus for maintenance immunosuppression. Importantly, there is an FDA black box warning against using sirolimus in conjunction with higher-dose CNIs due to higher risk of mortality and graft loss in liver transplantation, as well as an increased risk of hepatic artery thrombosis within the first month after transplant.
Everolimus is another mTOR inhibitor that has recently been approved for use in kidney and liver transplantation. Everolimus is a hydroxylated derivative of sirolimus, and its preserved structure allows for a similar mechanism of action via FKBP12 binding and mTORC1 inhibition. Its immediate appeal was its lower risk of nephrotoxicity. In a recent multicenter prospective clinical trial, patients with reduced-dose tacrolimus and everolimus had improved GFR at 2 years posttransplant compared to patients on standard tacrolimus-based regimens, without any higher risk of biopsy-proved acute rejection or major side effects.72,73 This improvement in GFR has also been identified retrospectively in patients converted to everolimus in maintenance immunosuppression.74
Side Effects of mTOR Inhibitors
Sirolimus has been associated with impaired wound healing after transplant, including anastomotic dehiscence in lung transplantation, surgical site infections, and fascial union problems.75–80 Surgeons confronted with transplant patients who are on mTOR inhibitors should consult with transplant professionals prior to operative intervention if possible, in order to guide alterations in immunosuppressive therapy to minimize complications. Several other complications have been noted in patients on mTOR inhibitors. Pulmonary toxicity manifested as pneumonitis has been identified, and caution should be taken in administering the drug to patients with pre-existing lung disease. Metabolic complications including hyperlipidemia and new-onset diabetes mellitus after transplant have also been identified as significant side effects.81 In low- to moderate-risk renal transplant recipients, conversion from tacrolimus to mTOR inhibitors was associated with a higher risk of acute rejection, higher rates of proteinuria, hypercholesterolemia, and anemia.81
Antimetabolite Drugs
Antimetabolite drugs in solid organ transplantation comprise a class of agents aimed at suppressing lymphocyte proliferation by reducing the rate of de novo purine synthesis, which prevents effective nucleotide synthesis and DNA transcription. Typically, these drugs are used in combination with CNIs and steroids. One of the first antimetabolite drugs applied in clinical transplantation was azathioprine. Azathioprine has been applied in a multitude of disease states including inflammatory bowel disease and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus. While it is effective in reducing T-cell and B-cell propagation, azathioprine is also a potent suppressive agent of bone marrow, increases the risk of anemia, and is also associated with pancreatitis.
A newer antimetabolite, mycophenolic acid (MPA), has emerged as a mainstay in solid organ transplantation. MPA was identified in a Penicillium fungal species, and is hepatically metabolized. MPA has less myelosuppressive activity than azathioprine, but side effects are common.
Side Effects of Antimetabolite Drugs
Myelosuppression and leukopenia are common side effects of this class of agents. Mycophenolate is also associated with a significant risk of gastrointestinal dysfunction including diarrhea, nausea, and vomiting. Azathioprine has been recognized as a group 1 carcinogen by the National Toxicology Program in the United States, related to length of use and dosage.82–85 Mycophenolate should be avoided in pregnancy or in transplant recipients trying to conceive due to the risk of miscarriage and congenital malformations.86–90
Steroids
Steroids have represented the mainstay of immunosuppressive therapy since the beginning of clinical transplantation. Glucocorticoids are steroids that arise from the zona fasciculata in the adrenal gland. These molecules have a plethora of effects in humans, and their receptors are omnipresent across human cell types. In lymphocytes, glucocorticoids act on cytosolic receptors that in turn lead to a variety of intracellular signals that are responsible for gene transcription, and decrease the expression of several cytokines, including IL-2. Glucocorticoids also reduce the number and function of both T cells and B cells.
Side Effects of Steroid Therapy
Steroid therapy is used in a variety of clinical treatments including maintenance immunosuppression and in high doses as a rescue therapy in patients presenting with acute rejection. There are several side effects to chronic corticosteroid use, which have prompted transplant clinicians and researchers to develop protocols that reduce the need for chronic steroid use. Common side effects include glucose intolerance, weight gain, skin and tissue frailty, osteopenia and osteoporosis, muscle breakdown, neuropsychiatric dysfunction, glaucoma and cataracts, secondary hypercortisolemia, and other effects. Many transplant programs are shifting toward steroid avoidance in maintenance immunosuppression in low-risk patients.91–93
TREATING REJECTION IN SOLID ORGAN TRANSPLANTATION
6 Immunologic rejection can occur minutes to years after an organ transplant. The early phases of rejection, termed hyperacute rejection, can occur on the operating room table to 24 hours after surgery and is mediated by the humoral immune system. Acute rejection is a cell-mediated phenomenon which often manifests days to months after transplant. Chronic rejection occurs in the months to years after transplant and continues to be one of the leading causes of graft loss in the long term. Acute and chronic antibody-mediated rejection (AMR) is a separate phenomenon that may occur days to years after transplant and remains an important barrier to successful engraftment of organs.
Hyperacute Rejection
Hyperacute rejection is a rare clinical entity in modern clinical transplantation. Hyperacute rejection is generally attributed to ABO blood group incompatibility or the presence of preformed anti-HLA antibodies. In the allograft, the capillary endothelium expresses blood group antigens which may join with circulating antibodies to A and B glycoproteins. For example, a blood group B transplant candidate would have circulating anti-A IgG antibodies in blood. If a blood group A kidney allograft were transplanted into that recipient without any other treatment, these antibodies would react with the donor endothelium and activate complement and humoral immunity effector mechanisms, which would lead to endothelial injury, thrombosis, as well as intraparenchymal hemorrhage. This response usually happens within minutes after reperfusion of the allograft, and requires removal of the allograft in order to prevent a systemic inflammatory response which may lead to multiorgan failure. Prevention of this type of rejection is central to the practice of clinical transplantation, and mandates confirmation of donor and recipient compatibility via ABO blood group verification and cross-matching prior to implantation of the allograft.
Acute Rejection
Induction protocols in combination with immunosuppressive therapy successfully prevent the relatively quick onset of acute rejection episodes. In the case of renal transplantation, induction therapies in combination with potent maintenance therapies have fostered a significant improvement of 1-year outcomes.94 Less than 10% of organs experience clinically significant acute cellular rejection (ACR) episodes leading to graft loss at 1 year.95 As discussed above, ACR is an adaptive response characterized by T-cell interactions with donor antigens either presented directly by the donor APC or indirectly by the recipient professional presenting cells.96 Once clonal expansion occurs “effector” T cells then infiltrate the graft and mediate various methods of organ injury.
The gold standard in pathologic categorization of renal transplant rejection is the Banff classification model. The Banff classification originated at a consensus conference in 1991 aimed at ranking the levels of rejection seen on kidney transplant biopsies based on degree of structural damage, such as tubulitis, arteritis, and cellular infiltrate.97 In renal transplantation, the diagnosis of ACR occurs with tissue confirmation after ruling other causes for declining graft function. Treatment of ACR is usually dictated by the Banff classification. For example, borderline rejection characterized by Banff class IA or IB is often treated with pulse corticosteroids and increases in maintenance immunosuppression whereas, more severe ACR is more often treated with multiple doses of thymoglobulin. Today, ACR is often the result of inadequate immunosuppression due to noncompliance/adherence or early immunosuppression withdrawal/underdosing. Repeated episodes of acute rejection are associated with chronic dysfunction and early graft failure.
Chronic Rejection
The clinical sequelae of chronic rejection (more recently termed chronic transplant dysfunction or CTD) are varied and organ specific. The unifying factor of CTD among all organs transplanted is the histopathologic effect of arterial intimal thickening in the vessels supplying the organ, termed transplant arteriosclerosis or transplant vasculopathy. Both immune and nonimmune stimuli may up-regulate pathways leading to progressive graft fibrosis. Immune-dependent mechanisms, including HLA mismatching, inadequate immunosuppression, and episodes of acute rejection, promote the development of CTD and eventual graft failure. Additionally, immune-independent mechanisms, such as prolonged ischemia time and usage of brain dead donor organs, as well as recipient factors such as native hypertension may also play a role in the onset of CTD. Various studies suggest that the development of vascular fibrosis progressing to parenchymal fibrosis is multifactorial and includes effector responses mediated by complement, CD4+ T cells, CD8+ T cells, and alloreactive antibodies.98–102
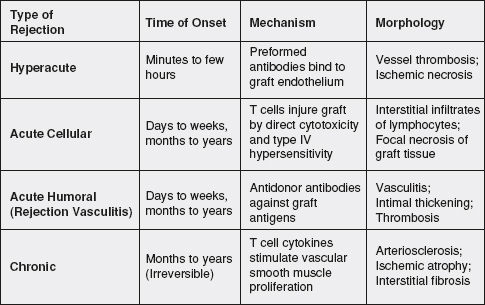
Figure 34-2. Time course, mechanism, and morphology of transplant rejection. Allograft rejection responses have many different phenotypes, which also lead to different therapies. This figure provides a summary of the types of clinically significant rejection episodes, the root cause behind them, and histopathologic features identified on biopsy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


