Figure 74.1. Clinical manifestations of cirrhosis.
COMPLICATIONS OF CIRRHOSIS
ASCITES
Ascites is defined as an accumulation of excessive fluid within the peritoneal cavity. Physiologically it reflects an excess of total-body sodium and water, either as a consequence of perceived underfilling, actual overfilling, or a combination of the two with some element of vasodilation. Ascites may be a complication of both hepatic and nonhepatic diseases. The most common causes of ascites in North America and Europe are shown in box 74.2.
Box 74.2 CAUSES OF ASCITES
| Cirrhosis | 85% |
| Mixed | 8% |
| Heart failure | 3% |
| Malignancy | 2% |
| Tuberculosis | <1% |
| Pancreatic | <1% |
| Nephrotic | <1% |
Diagnostic Evaluation of Ascites
The key steps in the evaluation of ascites include acquiring a thorough history and physical, obtaining an ascitic fluid evaluation, and obtaining special tests. Clinically, ascites is suggested by the presence of abdominal distension, bulging flanks, shifting dullness, and elicitation of a “puddle sign” in patients in the knee-elbow position. A fluid wave may be elicited in patients with massive tense ascites. Ascites may be graded as follows: grade 1, mild, only visible on ultrasound; grade 2, detectable with flank bulging and shifting dullness; and grade 3, directly visible, confirmed with fluid thrill. On physical examination the presence of vascular spiders, abdominal wall collaterals, and an umbilical nodule are useful in supporting the diagnosis of chronic liver disease as a cause of ascites.
Paracentesis is routine for new-onset ascites in patients on admission and in patients who have a clinical deterioration. A diagnostic tap is performed with a 22-gauge 1.5-inch needle, whereas a therapeutic tap is performed with a 15- to 18-gauge needle. There is an approximate 1% complication rate with paracentesis. A popular option for the tap site is the left lower quadrant of the abdomen, two fingerbreadths medial and cephalad to the anterior superior iliac spine. Tests are in table 74.1.
Table 74.1 ASCITES TESTS
| ROUTINE | OPTIONAL | SPECIAL |
| Cell count | Glucose | Cytology |
| Albumin | Lactate dehydrogenase | TB smear and culture |
| Culture | Gram stain | Triglycerides |
| Total protein | Bilirubin | |
| Amylase | ||
Currently the best way to classify ascites is to base it on the serum-ascites albumin gradient (SAAG). The SAAG is calculated by subtracting the ascitic fluid albumin value from the serum albumin value; it correlates directly with portal pressure. The specimens should be obtained relatively simultaneously. The accuracy of the SAAG results is approximately 97% in classifying ascites. Ascites with a SAAG gradient of >1.1 g/dL is termed high-albumin-gradient or portal hypertensive ascites. In contrast, ascites with a SAAG gradient of <1.1 g/dL is known as low-albumin-gradient or nonportal hypertensive and is associated with peritoneal diseases (table 74.2). In the past, ascites was classified as transudative or exudative based on the ascitic protein level (exudative if ascites protein was >2.5 g/dL), but this is no longer a favored method of classification. Classic causes of transudative ascites are portal hypertension secondary to cirrhosis and congestive heart failure. Examples of exudative ascites include peritoneal carcinomatosis and tuberculous peritonitis.
Table 74.2 SERUM ASCITES ALBUMIN GRADIENT
| HIGH (> 1.1 g/dL) | LOW (<1.1 g/dL) |
| Cirrhosis | Peritoneal cancer |
| Alcoholic hepatitis | Tuberculosis |
| Congestive heart failure | Pancreatitis |
| Massive liver metastates | Bile leak |
| Fulminant liver failure | Nephrotic syndrome |
| Budd-Chiari syndrome | Lupus serositis |
SOURCE: Reprinted with permission from Runyon BA, Montano AA, Antillon MR, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215–220.
Chylous ascites, caused by obstruction of the thoracic duct or cisterna chyli, most often is due to malignancy (e.g., lymphoma), but occasionally is observed postoperatively and following radiation injury. Chylous ascites also may be observed in the setting of cirrhosis. The ascites triglyceride concentration is >110 mg/dL. In addition, ascites triglyceride concentrations are greater than those observed in plasma. Ascitic fluid with >250 polymorphonuclear (PMN) cells/mm3 defines neutrocytic ascites and SBP. Many cases of ascites fluid with >1000 PMN/mm3 (and certainly >5000 PMN/mm3) are associated with appendicitis or a perforated viscus with resulting bacterial peritonitis. Appropriate radiologic studies must be performed in such patients to rule out surgical causes of peritonitis. Lymphocyte-predominant ascites raises concerns about the possibility of underlying malignancy or tuberculosis. Similarly, grossly bloody ascites may be observed in malignancy and tuberculosis. Bloody ascites is seen infrequently in uncomplicated cirrhosis. A common clinical dilemma is how to interpret the ascites PMN count in the setting of bloody ascites. We recommend subtraction of 1 PMN for every 250 RBCs in ascites to ascertain a corrected PMN count.
Special testing includes an abdominal ultrasound (ultrasound with Doppler can help assess the patency of hepatic vessels), cytology for peritoneal cancer (often requires a large volume for preparation of a cell block), cardiac echo for suspected cardiac ascites, and tuberculosis (TB) smear and culture. Upper GI endoscopy for screening of large varices is also an option. Factors associated with worsening of ascites include excess fluid or salt intake, malignancy, venous occlusion (e.g., Budd-Chiari syndrome), progressive liver disease, and spontaneous bacterial peritonitis (SBP).
Treatment of Ascites
Therapy for ascites should be tailored to the patient’s needs. Options are shown in table 74.3. Some patients with mild ascites respond to sodium restriction or diuretics taken once or twice per week. Other patients require aggressive diuretic therapy, careful monitoring of electrolytes, and occasional hospitalization to facilitate even more intensive diuresis. The development of massive ascites that is refractory to medical therapy has dire prognostic implications, with only 50% of patients surviving 6 months.
Table 74.3 TREATMENT OPTIONS FOR DIURETIC-RESPONSIVE AND DIURETIC-RESISTANT ASCITES
| CONVENTIONAL TREATMENT OPTIONS | KEY FEATURES |
| Abstinence from Alcohol | |
| Sodium restriction | First line of therapy |
| Dietary sodium restriction <2000 mg sodium per day and for refractory ascites as much as <500 mg/day | |
| Diuretics | Second line therapy |
| Spironolactone (Aldactone) blocks the aldosterone receptor at the distal tubule. Dose 50–300 mg once per day. Alternative is eplerenone | |
| Furosemide (Lasix) may be used as a solo agent or in combination with spironolactone. Furosemide blocks sodium reuptake in the loop of Henle. Dosed at 40–240 mg/day in 1–2 divided doses. Avoid intravenous furosemide if possible because it may precipitate acute kidney injury (AKI). | |
| Starting doses: 100 mg/day spironolactone and 40 mg/day furosemide | |
| Options for Diuretic-Resistant Ascites | |
| Large-volume paracentesis | Indicated when aggressive diuretic therapy is ineffective in controlling ascites (~5–10% of patients). |
| Several large randomized, controlled trials have shown that repeated large-volume paracentesis (4–6 L) is safer and more effective for the treatment of tense ascites compared with larger-than-usual doses of diuretics. | |
| Procedure-associated risks include a 1% chance of significant abdominal-wall hematoma, 0.01% chance of hemoperitoneum, and a 0.01% chance of iatrogenic infection related to paracentesis. The only absolute contraindication to paracentesis is clinically evident fibrinolysis and disseminated intravascular coagulation. | |
| Controversy around whether to use salt-poor albumin (SPA) with a tap. One option is to reserve SPA for taps >5 L. | |
| Vasopressin V2 receptor antagonist, e.g., Satavaptan | Improve diuresis and decrease the need for paracentesis in patients with diuretic-refractory ascites |
| Transjugular intrahepatic portosystemic shunt (TIPS) | A flexible metal prosthesis is used to bridge a branch of the hepatic and portal veins and is effective in reducing sinusoidal pressure. |
| The procedure is performed percutaneously under radiologic guidance and obviates the need for surgery. | |
| It is recommended that coagulopathy (INR > 2 and platelet count < 50 × 109/L) be corrected first if indicated and that paracentesis be performed in patients with tense ascites prior to the procedure. | |
| Four randomized, controlled studies have compared TIPS with large-volume paracentesis in refractory ascites. All 4 studies showed better control of ascites with TIPS, but only one study showed a survival benefit. | |
| The rate of procedure-related complications is 10%, and of procedure-related mortality is 2%. | |
| Procedure-related complications include neck hematomas, hemobilia, puncture of the liver capsule causing intra-abdominal bleeding, and shunt occlusion. | |
| Absolute contraindications for TIPS insertion include serum bilirubin > 85 µmol/L (5 mg/dL), INR >2, functional renal disorder with serum creatinine >250 µmol/L (2.8 mg/dL), intrinsic renal disease with urine protein >500 mg/24 hours or active urinary sediment, grade III or IV hepatic encephalopathy, cardiac disease, portal vein thrombosis, noncompliance with sodium restriction, or the presence of carcinoma that is likely to limit the patient’s lifespan to <1 year. Relative contraindications include dental sepsis, spontaneous bacterial peritonitis, and active infection (pneumonia or urinary tract infection). | |
| Liver transplantation | Liver transplantation is the only definitive treatment for ascites and the only treatment that has been clearly shown to improve survival. |
| Patients with cirrhosis who develop ascites should be assessed for possible liver transplantation because of their poor prognosis. | |
| Patients who develop renal dysfunction (GFR <50 mL/min) do much worse after liver transplantation (80% vs. 50% survival at 15 months) | |
| Other poor prognostic indicators include mean arterial pressure <82 mm Hg, urinary sodium excretion of <1.5 mEq/day, plasma norepinephrine levels of >570 pg/mL, poor nutritional state, presence of hepatomegaly, and serum albumin <25 g/L. | |
Complications of Ascites
Spontaneous Bacterial Peritonitis
SBP is observed in 15–26% of patients hospitalized with ascites. The syndrome arises most commonly in patients whose low-protein ascites (<1 g/dL) contains low levels of complement resulting in decreased opsonic activity. The three most common signs of SBP—abdominal pain, fever, and leukocytosis—are seen in only 70% of persons with SBP, and some argue that paracentesis should be performed in all patients with cirrhosis who have ascites at the time of hospitalization. SBP appears to be caused by the translocation of GI tract bacteria across the gut wall and also by the hematogenous spread of bacteria. The most common causative organisms are Escherichia coli, Streptococcus pneumoniae, Klebsiella species, and other gram-negative enteric organisms.
Classic SBP is diagnosed by the presence of neutrocytosis, which is defined as >250 PMN cells/mm3 of ascites in the setting of a positive ascites culture. Culture-negative neutrocytic ascites is observed more commonly. The yield of ascites culture studies may be increased by directly inoculating 10 mL of ascites into aerobic and anaerobic culture bottles at the patient’s bedside. Both conditions represent serious infections that carry a 20–30% mortality rate.
The most commonly used regimen in the treatment of SBP is a 5-day course of cefotaxime at 1–2 g intravenously every 8 hours. Alternatives include oral ofloxacin and other intravenous antibiotics with activity against gram-negative enteric organisms. Many authorities advise repeat paracentesis in 48–72 hours to document a decrease in the ascites PMN count to <250 cells/mm3 and to ensure the efficacy of therapy.
Once SBP develops, patients have a 70% chance of redeveloping the condition within 1 year. Prophylactic antibiotic therapy can reduce the recurrence rate of SBP to 20%. Some of the regimens used in the prophylaxis of SBP include norfloxacin at 400 mg orally every day 12 and trimethoprim-sulfamethoxazole at one double-strength tablet 5 days per week.
Therapy with norfloxacin at 400 mg orally twice per day for 7 days can reduce serious bacterial infection in patients with cirrhosis who have GI bleeding. Furthermore, it can be argued that all patients with low-protein ascites should undergo prophylactic therapy (e.g., with norfloxacin 400 mg/day orally) at the time of hospital admission given the high incidence of hospital-acquired SBP.
Massive Ascites
Patients with massive ascites may experience abdominal discomfort, depressed appetite, and decreased oral intake. Diaphragmatic elevation may lead to symptoms of dyspnea. Pleural effusions may result from the passage of ascitic fluid across channels in the diaphragm.
Umbilical and inguinal hernias are common in patients with moderate and massive ascites. The use of an elastic abdominal binder may protect the skin overlying a protruding umbilical hernia from maceration and may help prevent rupture and subsequent infection. Timely large-volume paracentesis also may help to prevent this disastrous complication. Umbilical hernias should not undergo elective repair unless patients are significantly symptomatic or their hernias are irreducible. As with all other surgeries in patients with cirrhosis, herniorrhaphy carries multiple potential risks, such as intraoperative bleeding, postoperative infection, and liver failure because of anesthesia-induced reductions in hepatic blood flow. However, these risks become acceptable in patients with severe symptoms from their hernia. Urgent surgery is necessary in the patient whose hernia has been complicated by bowel incarceration.
PORTAL HYPERTENSION
The normal liver has the ability to accommodate large changes in portal blood flow without appreciable alterations in portal pressure. Portal hypertension results from a combination of increased portal venous inflow and increased resistance to portal blood.
The portal hypertension of cirrhosis is caused by the disruption of hepatic sinusoids. However, portal hypertension may be observed in a variety of noncirrhotic conditions. Prehepatic causes include splenic vein thrombosis and portal vein thrombosis. These conditions commonly are associated with hypercoagulable states and with malignancy (e.g., pancreatic cancer).
Intrahepatic causes of portal hypertension are divided into presinusoidal, sinusoidal, and postsinusoidal conditions. The classic form of presinusoidal disease is caused by the deposition of Schistosoma oocytes in presinusoidal portal venules, with the subsequent development of granulomata and portal fibrosis. Schistosomiasis is the most common noncirrhotic cause of variceal bleeding worldwide. Schistosoma mansoni infection is described in Puerto Rico, Central and South America, the Middle East, and Africa. Schistosoma japonicum is described in the Far East. Schistosoma hematobium, observed in the Middle East and Africa, can produce portal fibrosis, but more commonly it is associated with urinary tract deposition of eggs. The classic sinusoidal cause of portal hypertension is cirrhosis. The classic postsinusoidal condition is an entity known as veno-occlusive disease. Obliteration of the terminal hepatic venules may result from ingestion of pyrrolizidine alkaloids in comfrey tea or Jamaican bush tea and following the high-dose chemotherapy that precedes bone marrow transplantation.
Posthepatic causes of portal hypertension may include chronic right-sided heart failure, tricuspid regurgitation, and obstructing lesions of the hepatic veins and inferior vena cava. These latter conditions, and the symptoms they produce, are termed Budd-Chiari syndrome. Predisposing conditions include hypercoagulable states, tumor invasion into the hepatic vein or inferior vena cava, and membranous obstruction of the inferior vena cava. Inferior vena cava webs are observed most commonly in South and East Asia and are postulated to be due to nutritional factors.
Symptoms of the Budd-Chiari syndrome are attributed to decreased outflow of blood from the liver, with resulting hepatic congestion and portal hypertension. These symptoms include hepatomegaly, abdominal pain, and ascites. Cirrhosis only ensues later in the course of disease. Differentiating the Budd-Chiari syndrome from cirrhosis by history or physical examination may be difficult. Thus, the Budd-Chiari syndrome must be included in the differential diagnosis of conditions that produce ascites and varices. A possible clue may come from the analysis of the ascitic fluid. The SAAG is usually >1.1, but the ascitic fluid has a high protein content unlike that of cirrhotic ascites. Hepatic vein patency is checked most readily by performing an abdominal ultrasound with Doppler examination of the hepatic vessels. Abdominal computed tomography (CT) scan with intravenous contrast, abdominal magnetic resonance imaging (MRI), and visceral angiography also may provide information regarding the patency of hepatic vessels.
HEPATORENAL SYNDROME
This syndrome represents a continuum of renal dysfunction that may be observed in patients with cirrhosis and is caused by the vasoconstriction of large and small renal arteries and the impaired renal perfusion that results. The syndrome may represent an imbalance between renal vasoconstrictors and vasodilators. Plasma levels of a number of vasoconstricting substances are elevated in patients with cirrhosis and include angiotensin, antidiuretic hormone, and norepinephrine. Renal perfusion appears to be protected by vasodilators, including prostaglandins E2 and I2 and atrial natriuretic factor. Nonsteroidal antiinflammatory drugs (NSAIDs) inhibit prostaglandin synthesis. They may potentiate renal vasoconstriction, with a resulting drop in glomerular filtration. Thus, the use of NSAIDs is contraindicated in patients with decompensated cirrhosis.
Most patients with hepatorenal syndrome are noted to have minimal histological changes in the kidneys. Kidney function usually recovers when patients with cirrhosis and hepatorenal syndrome undergo liver transplantation. In fact, a kidney donated by a patient dying from hepatorenal syndrome functions normally when transplanted into a renal transplant recipient.
Hepatorenal syndrome progression may be slow (type II) or rapid (type I). Type I disease frequently is accompanied by rapidly progressive liver failure. Hemodialysis offers temporary support for such patients. These individuals are salvaged only by performance of liver transplantation. Exceptions to this rule are the patients with FHF or severe alcoholic hepatitis, who spontaneously recover both liver and kidney function. In type II hepatorenal syndrome patients may have stable or slowly progressive renal insufficiency. Many such patients develop ascites that is resistant to management with diuretics.
Hepatorenal syndrome is diagnosed when a creatinine clearance <40 mL/min is present or when the serum creatinine >1.5 mg/dL, urine volume <500 mL/day, and urine sodium <10 mEq/L are present. Urine osmolality is greater than plasma osmolality. In hepatorenal syndrome, renal dysfunction cannot be explained by pre-existing kidney disease, prerenal azotemia, the use of diuretics, or exposure to nephrotoxins. Clinically, the diagnosis may be reached if central venous pressure is determined to be normal or if no improvement of renal function occurs following the infusion of at least 1.5 L of a plasma expander.
Nephrotoxic medications, including aminoglycoside antibiotics, should be avoided in patients with cirrhosis. Patients with early hepatorenal syndrome may be salvaged by aggressive expansion of intravascular volume with albumin and fresh frozen plasma and by avoidance of diuretics. Administration of oral prostaglandins may be beneficial, but this point is controversial. Use of renal-dose dopamine is not effective.
HEPATIC ENCEPHALOPATHY
Hepatic encephalopathy is a syndrome observed in some patients with cirrhosis that is marked by personality changes, intellectual impairment, and a depressed level of consciousness. The diversion of portal blood into the systemic circulation appears to be a prerequisite for the syndrome. Indeed, hepatic encephalopathy may develop in patients who do not have cirrhosis who undergo portocaval shunt surgery.
Clinical Features of Hepatic Encephalopathy
The symptoms of hepatic encephalopathy may range from mild to severe and may be observed in as many as 70% of patients with cirrhosis. Symptoms are graded on the following scale:
Grade 0—Subclinical; normal mental status, but minimal changes in memory, concentration, intellectual function, coordination
Grade 1—Mild confusion, euphoria or depression, decreased attention, slowing of ability to perform mental tasks, irritability, disorder of sleep pattern (i.e., inverted sleep cycle)
Grade 2—Drowsiness, lethargy, gross deficits in ability to perform mental tasks, obvious personality changes, inappropriate behavior, intermittent disorientation (usually to time)
Grade 3—Somnolent but arousable, unable to perform mental tasks, disorientation to time and place, marked confusion, amnesia, occasional fits of rage, speech is present but incomprehensible
Grade 4—Coma, with or without response to painful stimuli
Patients with mild and moderate hepatic encephalopathy demonstrate decreased short-term memory and concentration on mental status testing. Findings on physical examination include asterixis and fetor hepaticus.
Laboratory Abnormalities in Hepatic Encephalopathy
An elevated arterial or free venous serum ammonia level is the classic laboratory abnormality reported in patients with hepatic encephalopathy. This finding may aid in the assignment of a correct diagnosis to a patient with cirrhosis who presents with altered mental status. However, serial ammonia measurements are inferior to clinical assessment in gauging improvement or deterioration in patients under therapy for hepatic encephalopathy. No utility exists for checking the ammonia level in a patient with cirrhosis who does not have evidence of encephalopathy.
Some patients with hepatic encephalopathy have the classic but nonspecific electroencephalogram (EEG) changes of high-amplitude low-frequency waves and triphasic waves. EEG may be helpful in the initial workup of a patient with cirrhosis and altered mental status when ruling out seizure activity may be necessary.
CT scan and MRI studies of the brain may be important in ruling out intracranial lesions when the diagnosis of hepatic encephalopathy is in question.
Common Precipitants of Hepatic Encephalopathy
Some patients with a history of hepatic encephalopathy may have normal mental status when under medical therapy. Others have chronic memory impairment in spite of medical management. Both groups of patients are subject to episodes of worsened encephalopathy. Common precipitants of hyperammonemia and worsening mental status are diuretic therapy, renal failure, GI bleeding, infection, and constipation. Dietary protein overload is an infrequent cause of worsening encephalopathy. Medications, notably opiates, benzodiazepines, antidepressants, and antipsychotic agents, also may worsen encephalopathy symptoms.
Nonhepatic causes of altered mental function must be excluded in patients with cirrhosis who have worsening mental function. A check of the blood ammonia level may be helpful in such patients. Precipitants of hepatic encephalopathy should be corrected (e.g., metabolic disturbances, GI bleeding, infection, and constipation).
Treatment of Hepatic Encephalopathy
Lactulose is helpful in patients with the acute onset of severe encephalopathy symptoms and in patients with milder, chronic symptoms. This nonabsorbable disaccharide stimulates the passage of ammonia from tissues into the gut lumen and inhibits intestinal ammonia production. Initial lactulose dosing is 30 mL orally once or twice daily. Dosing is increased until the patient has 2–4 loose stools per day. Dosing should be reduced if the patient complains of diarrhea, abdominal cramping, or bloating. Higher doses of lactulose may be administered via either a nasogastric tube or rectal tube to hospitalized patients with severe encephalopathy. Other cathartics, including colonic lavage solutions, that contain polyethylene glycol (PEG) (e.g., Go-Lytely) also may be effective in patients with severe encephalopathy.
Neomycin and other antibiotics (e.g., metronidazole, oral vancomycin, paromomycin, oral quinolones) serve as second-line agents. They work by decreasing the colonic concentration of ammoniagenic bacteria. Neomycin dosing is 250–1000 mg orally two to four times daily. Treatment with neomycin may be complicated by ototoxicity and nephrotoxicity.
Rifaximin is a nonabsorbable antibiotic that received approval by the US Food and Drug Administration (FDA) in 2004 for the treatment of travelers’ diarrhea. Experience in Europe over the last two decades suggests that rifaximin can decrease colonic levels of ammoniagenic bacteria with resulting improvement in hepatic encephalopathy symptoms. Typical rifaximin dosing in European hepatic encephalopathy trials was two 200mg tablets taken orally three times daily. Work is being done to determine if lower doses of the medication can effectively treat hepatic encephalopathy. One meta-analysis has suggested that rifaximin may be more effective than lactulose in the treatment of hepatic encephalopathy.
Other chemicals capable of decreasing blood ammonia levels are l-ornithine-l-aspartate (available in Europe) and sodium benzoate.
Low-protein diets were recommended routinely in the past for patients with cirrhosis. High levels of aromatic amino acids contained in animal proteins were believed to lead to increased blood levels of the false neurotransmitters tyramine and octopamine, with resulting worsening of encephalopathy symptoms. In our experience, the vast majority of patients can tolerate a protein-rich diet (>1.2 g/kg/day) including well-cooked chicken, fish, vegetable protein, and, if needed, protein supplements.
Protein restriction is rarely necessary in patients with chronic encephalopathy symptoms. Many patients with cirrhosis have protein-calorie malnutrition at baseline.
PULMONARY ABNORMALITIES IN CIRRHOSIS
Hepatopulmonary Syndrome
In hepatopulmonary syndrome (HPS), pulmonary arteriovenous anastomoses result in arteriovenous shunting. HPS is characterized by the symptom of platypnea (dyspnea that is relieved when lying down and worsens when sitting or standing up). HPS is a potentially progressive and life-threatening complication of cirrhosis that can be detected most readily by echocardiographic visualization of late-appearing bubbles in the left atrium following the injection of agitated saline. Patients can receive a diagnosis of HPS when their Pao2 is <70 mm Hg. Some cases of HPS may be corrected by liver transplantation.
Portopulmonary Hypertension
The etiology of portopulmonary hypertension (PPHTN) is unknown. It is defined as the presence of a mean pulmonary artery pressure >25 mm Hg in the setting of a normal pulmonary capillary wedge pressure. Patients who develop severe PPHTN may require aggressive medical therapy in an effort to stabilize pulmonary artery pressures and to decrease their chance of perioperative mortality in the setting of liver transplantation.
HEPATIC HYDROTHORAX
This condition results in the formation of pleural effusions in patients with cirrhosis. In most patients there is concurrent ascites. In all patients with cirrhosis, the presence of pleural effusions should induce a concern that liver disease is the major culprit. Effusions are often right-sided (85–90%) but can also be left-sided or bilateral. Therapy is geared toward the liver disease and management of ascites. Chest tubes are contraindicated.
HEPATOCELLULAR CANCER
Hepatocellular carcinoma (HCC) occurs in 10–25% of patients with cirrhosis in the United States and is most often associated with hemochromatosis, alpha-1-antitrypsin deficiency, hepatitis B, hepatitis C, and alcoholic cirrhosis. HCC is observed less commonly in primary biliary cirrhosis and is a rare complication of Wilson disease. Regardless, current guidelines suggest that all patients with cirrhosis irrespective of cause should be screened for HCC. Recommended screening is ultrasonography every 6–12 months. Cholangiocarcinoma occurs in approximately 10% of patients with primary sclerosing cholangitis some of whom may have cirrhosis as a result.
PROGNOSIS OF CIRRHOSIS
Gauging prognosis in cirrhosis is important. Two scoring systems have been used (table 74.4). These are the Child-Turcotte-Pugh (CTP) system and the Model for End-Stage Liver Disease (MELD) scoring system. Epidemiologic work shows that the CTP score may predict life expectancy in patients with advanced cirrhosis. A CTP score of 10 or greater is associated with a 50% chance of death within 1 year. The MELD scoring system has been used by liver transplant programs in the United States to assess the relative severities of patients’ liver diseases. MELD scores range from 6 to 40 points. The 3-month mortality statistics are associated with the following MELD scores: MELD score of <9, 2.9% mortality; MELD score of 10–19, 7.7% mortality; MELD score of 20–29, 23.5% mortality; MELD score of 30–39, 60% mortality; and MELD score of >40, 81% mortality.
Table 74.4 CHILD-TURCOTTE-PUGH SCORING SYSTEM FOR CIRRHOSIS
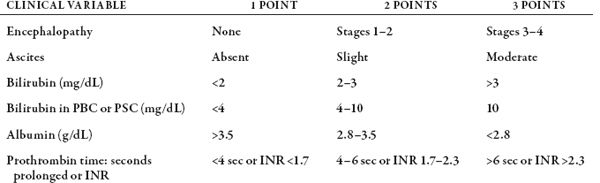
NOTE: Child class A = 5–6 points; Child class B = 7–9 points; Child class C = 10–15 points. SOURCE: Child CG, Turcotte JG. Surgery and portal hypertension. In Child Cg, ed. The Liver and Portal Hypertension. Philadelphia: Saunders; 1964:50–64.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


