Choline1
Steven H. Zeisel
1Abbreviations: AI, adequate intake; CDP-choline, cytidine diphosphocholine; CHDH, choline dehydrogenase; ERE, estrogen-response elements; LTP, long-term potentiation; MTHFR, methylenetetrahydrofolate reductase; PEMT, phosphatidylethanolamine N-methyltransferase; SNP, single nucleotide polymorphisms; SRE, sterol-responsive element; TPN, total parenteral nutrition; UL, tolerable upper intake level; VLDL, very-low-density lipoprotein.
Choline was discovered in 1862 and was chemically synthesized in 1866 (1), but it was not recognized as one of the nutrients required by humans until 1998 (2). The importance of choline as a nutrient in animals was first appreciated more than 50 years earlier, during the pioneering work on insulin (3). Depancreatized dogs, maintained on insulin, developed fatty infiltration of the liver and died. Administration of raw pancreas prevented fatty liver and hepatic damage; the active component was the choline moiety of pancreatic phosphatidylcholine (4). The recognition that choline was needed by humans took so long because, like vitamin D, the choline moiety can be produced endogenously (when phosphatidylcholine is formed from phosphatidylethanolamine, mainly in the liver). Investigators assumed that this biosynthesis could meet human needs, but this is not true in most men and postmenopausal women (5). The gene for the enzyme catalyzing this biosynthesis is induced by estrogen (6), and some young women may not need to eat choline (5). As discussed in more detail later, genetic variation also contributes to a wide variation in the dietary requirement for choline.
In 1998, the US Institute of Medicine’s Food and Nutrition Board established an adequate intake (AI) and tolerable upper intake limit (UL) for choline (Table 30.1) (2). The AI is approximately 550 mg/70 kg body weight, with more recommended during pregnancy and lactation. The AI for infants is estimated from the calculated intake from human breast milk. The UL for choline (see Table 30.1) was derived from the lowest-observed-adverse-effect level (hypotension) in humans and is 3 g/day for an adult (2). Human studies of choline requirements in children or infants have not been conducted. As discussed later, women need less dietary choline because of enhanced endogenous biosynthesis (5, 6), but pregnancy and lactation require large amounts of choline and likely increase the requirement for this nutrient (7).
Choline has several important functions: it is a source of methyl groups needed to make S-adenosylmethionine, it is a part of the neurotransmitter acetylcholine, and it is a part of the predominant phospholipids in membranes (phosphatidylcholine and sphingomyelin) (8). Betaine, formed from choline, is an important osmolyte in the kidney glomerulus and helps with the reabsorption of water from the kidney tubule (9). Although they represent a smaller proportion of the total choline pool, important metabolites of choline include platelet-activating factor, choline plasmalogens, lysophosphatidylcholine, phosphocholine, and glycerophosphocholine (8).
DIETARY SOURCES
Many foods eaten by humans contain significant amounts of choline and esters of choline (10, 11). Eggs and liver are excellent sources of choline; one egg contains approximately 33% of the daily requirement (see the US Department of Agriculture’s website for a list of dietary sources of choline and betaine: http://www.nal.usda.gov/fnic/foodcomp/Data/Choline/Choline.html). Humans on an ad libitum diet ingest between 150 and 600 mg choline/day (as free choline and choline esters) (12, 13, 14, 15, 16, 17). In the 2005 National Health and Nutrition Examination Survey, only a few participants in all age groups in the United States ate diets achieving the recommended intake for choline (˜550 mg/day/70 kg body
weight) (18). Foods also contain the choline metabolite betaine (10), which cannot be converted to choline but can be used as a methyl donor, thereby sparing some choline requirements (19). Plant-derived food sources can be a rich source of betaine (named after beets), but only membranerich plant components (e.g., wheat germ) contain significant amounts of choline.
weight) (18). Foods also contain the choline metabolite betaine (10), which cannot be converted to choline but can be used as a methyl donor, thereby sparing some choline requirements (19). Plant-derived food sources can be a rich source of betaine (named after beets), but only membranerich plant components (e.g., wheat germ) contain significant amounts of choline.
TABLE 30.1 DIETARY REFERENCE INTAKE VALUES FOR CHOLINE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Human breast milk is rich in choline compounds (20). The bioavailability of choline may differ between human milk and infant formulas (20, 21), because they contain different amounts of the various choline compounds. In 2007, most commercial infant formulas were modified to “humanize” their choline content to approximate the amount present in mature human breast milk. Where does all this choline in human milk come from? Mammary epithelial cells are capable of concentrative uptake of choline from maternal blood (22) and of the biosynthesis of choline de novo (23) through phosphatidylethanolamine N-methyltransferase (PEMT) activity; this is the only pathway for endogenous biosynthesis of the choline moiety. The free choline content of human milk is very high at the start of lactation and diminishes by 30 days postpartum (24). Breast milk phosphatidylcholine and plasma choline concentrations are influenced by dietary choline intake, and a dietary supplement of phosphatidylcholine can further increase breast milk choline, betaine, and phosphocholine concentrations (25).
DIGESTION AND ABSORPTION
The extent to which dietary choline is bioavailable depends on the efficiency of its absorption from the intestine. In adults, some ingested choline is metabolized before it can be absorbed from the gut. Gut bacteria degrade choline to form betaine and to make methylamines (26) and may destroy enough diet-derived choline to influence the human dietary requirement (27, 28). Some of the variation in human requirements may be caused by differences in the intestinal microbiome. The free choline surviving these fates is absorbed by the intestine by carrier-mediated transport (29, 30). At this time, no other component of the diet has been identified as competing with choline for transport by intestinal carriers. Both pancreatic secretions and intestinal mucosal cells contain enzymes (phospholipases A1, A2, and B) capable of hydrolyzing phosphatidylcholine in the diet. The free choline that is formed enters the portal circulation of the liver (31).
Large amounts of choline are delivered to the fetus across the placenta, where choline transport systems pump it against a concentration gradient (32). The placenta is one of the few nonnervous tissues to store large amounts of choline as acetylcholine (33). Perhaps this is a special reserve storage pool that ensures delivery of choline to the fetus. In utero, the fetus is exposed to very high choline concentrations, with a progressive decline in blood choline concentration thereafter until adult levels are achieved after the first weeks of life (34). In fact, plasma or serum choline concentrations are six to sevenfold higher in the fetus and newborn than they are in the adult (35, 36). High levels of choline circulating in the neonate presumably ensure enhanced availability of choline to tissues. Neonatal rat brain efficiently extracts choline from blood (37), and increased serum choline in the neonatal rat is associated with twofold higher choline concentration in neonatal brain than is present later in life. Supplementing choline during the perinatal period further increases choline metabolite concentrations in blood and brain (38).
All tissues accumulate choline by diffusion and mediated transport, but uptake by liver, kidney, mammary gland, placenta, and brain is especially important (30, 39). A specific carrier mechanism transports free choline across the blood-brain barrier at a rate that is proportional to serum choline concentration, and, in the neonate, this choline transporter has especially high capacity (37, 40). Hepatectomy increases the half-life of choline and results in an increase in blood choline concentration. The rate at which liver takes up choline is sufficient to explain the rapid disappearance of choline injected systemically.
The kidney also accumulates choline (41). Some of this choline appears in the urine unchanged, but most is oxidized within the kidney to form betaine (42, 43, 44, 45) and glycerophosphocholine (46). Both substances are important intracellular osmoprotectants within kidney. Mean free choline concentrations in the plasma of azotemic humans are several times greater than in normal controls (47). Hemodialysis rapidly removes choline from the plasma (48, 49). Renal transplantation in humans lowers plasma choline from 30 µM in the azotemic patient to 15 µM within 1 day (50).
The kidney also accumulates choline (41). Some of this choline appears in the urine unchanged, but most is oxidized within the kidney to form betaine (42, 43, 44, 45) and glycerophosphocholine (46). Both substances are important intracellular osmoprotectants within kidney. Mean free choline concentrations in the plasma of azotemic humans are several times greater than in normal controls (47). Hemodialysis rapidly removes choline from the plasma (48, 49). Renal transplantation in humans lowers plasma choline from 30 µM in the azotemic patient to 15 µM within 1 day (50).
METABOLISM
Only a small fraction of dietary choline is acetylated (Fig. 30.1), catalyzed by the activity of choline acetyltransferase (51). This enzyme is highly concentrated in the terminals of cholinergic neurons, but it is also present in such nonnervous tissues as the placenta. The availability of choline and acetyl-coenzyme A (CoA) influences choline acetyltransferase activity. In brain, it is unlikely that choline acetyltransferase is saturated with either of its substrates, so choline (and possibly acetyl-CoA) availability determines the rate of acetylcholine synthesis (51). Increased brain acetylcholine synthesis is associated with an augmented release into the synapse of this neurotransmitter (52, 53, 54). Choline taken up by brain may first enter a storage pool (perhaps the phosphatidylcholine in membranes) before it is converted to acetylcholine (55). The choline phospholipids in cholinergic neurons comprise a large precursor pool of choline available for use in acetylcholine synthesis (56). This feature may be especially important in neurons with increased demands for choline to sustain acetylcholine release (e.g., when particular cholinergic neurons fire frequently or when the supply of choline from the extracellular fluid is inadequate).
The methyl groups of choline can be made available from one-carbon metabolism, on conversion to betaine (8) (see Fig. 30.1). Formation of betaine involves oxidation to betaine aldehyde in the inner mitochondrial membrane (57, 58), followed by oxidation of betaine aldehyde (catalyzed by betaine aldehyde dehydrogenase or by a nonspecific aldehyde dehydrogenase in the mitochondria and in the cytosol) to form betaine. Liver and kidney are the major sites of choline oxidation. Betaine cannot be reduced back to choline. Thus, the oxidation pathway acts to diminish the availability of choline to tissues while at the same time scavenging some methyl groups. This pathway is also important for mitochondrial adenosine triphosphate (ATP) generation, because mice with the choline dehydrogenase (Chdh) gene knocked out have defective mitochondrial ATP production (58).
The demand for choline as a methyl-group donor is probably the major factor that determines how rapidly a diet deficient in choline will induce pathologic features. The metabolisms of choline, methionine, and methylfolate are closely interrelated (see Fig. 30.1). The pathways intersect at the formation of methionine from homocysteine. Betaine:homocysteine methyltransferase, a zinc metalloenzyme (59), catalyzes the methylation of homocysteine using the choline metabolite betaine as the methyl donor (59, 60). In an alternative pathway, 5-methyltetrahydrofolate:homocysteine methyltransferase regenerates methionine by using a methyl group derived de novo from the one-carbon pool (61). Perturbing the metabolism of one of the methyl donors results in compensatory changes in the other methyl donors because of the intermingling of these metabolic pathways (62, 63, 64, 65, 66, 67, 68).
Rats ingesting a choline-deficient diet showed diminished tissue concentrations of methionine and S-adenosylmethionine (66) and of total folate (63). Methotrexate, which is widely used in the treatment of cancer, psoriasis, and rheumatoid arthritis, limits the availability of methyl groups by competitively inhibiting dihydrofolate reductase, a key enzyme in intracellular folate metabolism. Rats treated with methotrexate have diminished pools of all choline metabolites in liver (69). Choline supplementation reverses the fatty liver caused by methotrexate administration (70, 71, 72, 73).
Genetically modified mice with defective methylene tetrahydrofolate reductase (MTHFR) activity become choline deficient (74). This observation is important because many humans have genetic polymorphisms that alter the activity of this enzyme (75, 76), and choline intake exceeding current dietary recommendations preserves markers of cellular methylation and attenuates DNA damage in men with the MTHFR C677T genotype (67).
The interrelationship between choline and folate is especially interesting because multiple studies in humans have demonstrated that individuals with diminished folate status are much more likely to have babies with neural tube defects (2, 77). In humans, women in the lowest quartile for dietary choline intake had an increased risk for having a baby with a neural tube defect or a cleft palate (15, 78). In mice, depletion of choline was associated with development of neural tube defects (79, 80). In addition, the intermingling of choline and homocysteine metabolism is important because increased plasma homocysteine concentration is an independent risk factor for cardiovascular disease (81). Homocysteine concentrations are lower when people eat diets higher in choline content (16, 82).
Elegant regulatory mechanisms control phosphatidylcholine biosynthesis and hydrolysis (83, 84). Synthesis occurs by two pathways (see Fig. 30.1). In the first pathway, choline is phosphorylated and then converted to cytidine diphosphocholine (CDP-choline). This high-energy intermediate, in combination with diacylglycerol, forms phosphatidylcholine and cytidine monophosphate. In the alternative pathway, phosphatidylethanolamine is sequentially methylated to form phosphatidylcholine, by using S-adenosylmethionine as the methyl donor.
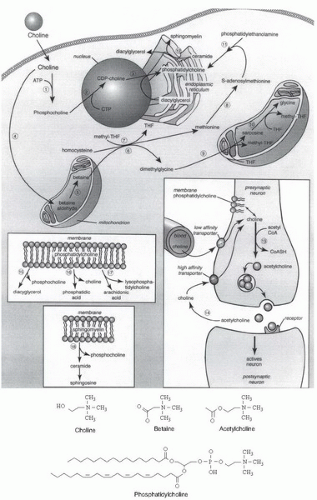 Fig. 30.1. Metabolism of choline. The three major metabolic uses for choline are as a precursor for phosphatidylcholine biosynthesis, as a methyl donor, and as a precursor for acetylcholine biosynthesis. ATP, adenosine triphosphate; CDP-choline, cytidine diphosphocholine; CoA, coenzyme A; CoASH, coenzyme A acetylcholine synthesis; CTP, cytidine triphosphate; THF, tetrahydrofolate. |
Choline kinase, the first enzyme in the CDP-choline pathway, has been purified, and its properties are reviewed elsewhere (85). This cytosolic enzyme also catalyzes the phosphorylation of ethanolamine. The next step in the pathway, catalyzed by CTP:phosphocholine cytidylyltransferase, is the rate-limiting and regulated step in phosphatidylcholine biosynthesis (86, 87). Deficient activity of this enzyme in the lungs of prematurely born human infants contributes to the respiratory distress syndrome (88). Cytidylyltransferase is present in the cytosol and in the nucleus (89) as an inactive dimer of two 42-kDa subunits and in the endoplasmic reticulum, Golgi apparatus, and nuclear envelope as an active membrane-bound form (83). Expression of the CTP:phosphocholine cytidylyltransferase gene is inhibited by a sterol-responsive element (SRE) in the promoter activated by cholesterol, 25-hydroxycholesterol, SREBP1a, or SREBP2 (90, 91).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


