26
Cholesterol Synthesis, Transport, & Excretion
Kathleen M. Botham, PhD, DSc & Peter A. Mayes, PhD, DSc
OBJECTIVES
After studying this chapter, you should be able to:
![]() Appreciate the importance of cholesterol as an essential structural component of cell membranes and as a precursor of all other steroids in the body, and indicate its pathological role in cholesterol gallstone disease and atherosclerosis development.
Appreciate the importance of cholesterol as an essential structural component of cell membranes and as a precursor of all other steroids in the body, and indicate its pathological role in cholesterol gallstone disease and atherosclerosis development.
![]() Identify the five stages in the biosynthesis of cholesterol from acetyl-CoA.
Identify the five stages in the biosynthesis of cholesterol from acetyl-CoA.
![]() Understand the role of 3-hydroxy-3-methylglutaryl CoA reductase (HMG-CoA reductase) in controlling the rate of cholesterol synthesis and explain the mechanisms by which its activity is regulated.
Understand the role of 3-hydroxy-3-methylglutaryl CoA reductase (HMG-CoA reductase) in controlling the rate of cholesterol synthesis and explain the mechanisms by which its activity is regulated.
![]() Appreciate that cholesterol balance in cells is tightly regulated and indicate the factors involved in maintaining the correct balance.
Appreciate that cholesterol balance in cells is tightly regulated and indicate the factors involved in maintaining the correct balance.
![]() Explain the role of plasma lipoproteins, including chylomicrons, very low density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL), in the transport of cholesterol between tissues in the plasma.
Explain the role of plasma lipoproteins, including chylomicrons, very low density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL), in the transport of cholesterol between tissues in the plasma.
![]() Name the two main primary bile acids found in mammals, outline the pathways by which they are synthesized from cholesterol in the liver, and understand the role of cholesterol 7a-hydroxylase in regulating the process.
Name the two main primary bile acids found in mammals, outline the pathways by which they are synthesized from cholesterol in the liver, and understand the role of cholesterol 7a-hydroxylase in regulating the process.
![]() Appreciate the importance of bile acid synthesis not only in the digestion and absorption of fats but also as a major excretory route for cholesterol.
Appreciate the importance of bile acid synthesis not only in the digestion and absorption of fats but also as a major excretory route for cholesterol.
![]() Indicate how secondary bile acids are produced from primary bile acids by intestinal bacteria.
Indicate how secondary bile acids are produced from primary bile acids by intestinal bacteria.
![]() Explain what is meant by the “enterohepatic circulation” and why it is important.
Explain what is meant by the “enterohepatic circulation” and why it is important.
![]() Identify the lifestyle factors that influence plasma cholesterol concentrations and thus affect the risk of coronary heart disease.
Identify the lifestyle factors that influence plasma cholesterol concentrations and thus affect the risk of coronary heart disease.
![]() Understand that the class of lipoprotein in which cholesterol is carried is important in determining the effects of plasma cholesterol on atherosclerosis development, with high levels of VLDL or LDL being deleterious and high levels of HDL being beneficial.
Understand that the class of lipoprotein in which cholesterol is carried is important in determining the effects of plasma cholesterol on atherosclerosis development, with high levels of VLDL or LDL being deleterious and high levels of HDL being beneficial.
![]() Give examples of inherited and noninherited conditions affecting lipoprotein metabolism that cause hypo- or hyperlipoproteinemia.
Give examples of inherited and noninherited conditions affecting lipoprotein metabolism that cause hypo- or hyperlipoproteinemia.
BIOMEDICAL IMPORTANCE
Cholesterol is present in tissues and in plasma either as free cholesterol or combined with a long-chain fatty acid as cholesteryl ester, the storage form. In plasma, both forms are transported in lipoproteins (Chapter 25). Cholesterol is an amphipathic lipid and as such is an essential structural component of membranes, where it is important for the maintenance of the correct permeability and fluidity, and of the outer layer of plasma lipoproteins. It is synthesized in many tissues from acetyl-CoA and is the precursor of all other steroids in the body, including corticosteroids, sex hormones, bile acids, and vitamin D. As a typical product of animal metabolism, cholesterol occurs in foods of animal origin such as egg yolk, meat, liver, and brain. Plasma low-density lipoprotein (LDL) is the vehicle that supplies cholesterol and cholesteryl ester to many tissues. Free cholesterol is removed from tissues by plasma high-density lipoprotein (HDL) and transported to the liver, where it is eliminated from the body either unchanged or after conversion to bile acids in the process known as reverse cholesterol transport (Chapter 25). Cholesterol is a major constituent of gallstones. However, its chief role in pathologic processes is as a factor in the genesis of atherosclerosis of vital arteries, causing cerebrovascular, coronary, and peripheral vascular disease.
CHOLESTEROL IS DERIVED ABOUT EQUALLY FROM THE DIET & FROM BIOSYNTHESIS
A little more than half the cholesterol of the body arises by synthesis (about 700 mg/d), and the remainder is provided by the average diet. The liver and intestine account for approximately 10% each of total synthesis in humans. Virtually all tissues containing nucleated cells are capable of cholesterol synthesis, which occurs in the endoplasmic reticulum and the cytosolic compartments.
Acetyl-CoA Is the Source of All Carbon Atoms in Cholesterol
The biosynthesis of cholesterol may be divided into five steps: (1) synthesis of mevalonate from acetyl-CoA (Figure 26–1); (2) formation of isoprenoid units from mevalonate by loss of CO2 (Figure 26–2); (3) condensation of six isoprenoid units form squalene (Figure 26–2); (4) cyclization of squalene give rise to the parent steroid, lanosterol. (5) formation of cholesterol from lanosterol (Figure 26–3).
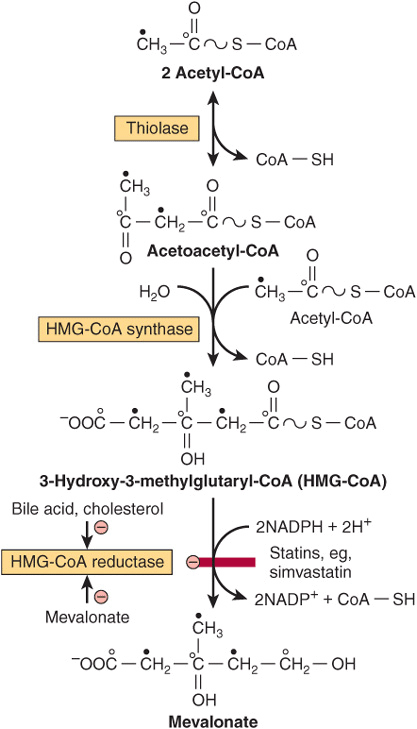
FIGURE 26–1 Biosynthesis of mevalonate. HMG-CoA reductase is inhibited by statins. The open and solid circles indicate the fate of each of the carbons in the acetyl moiety of acetyl-CoA.
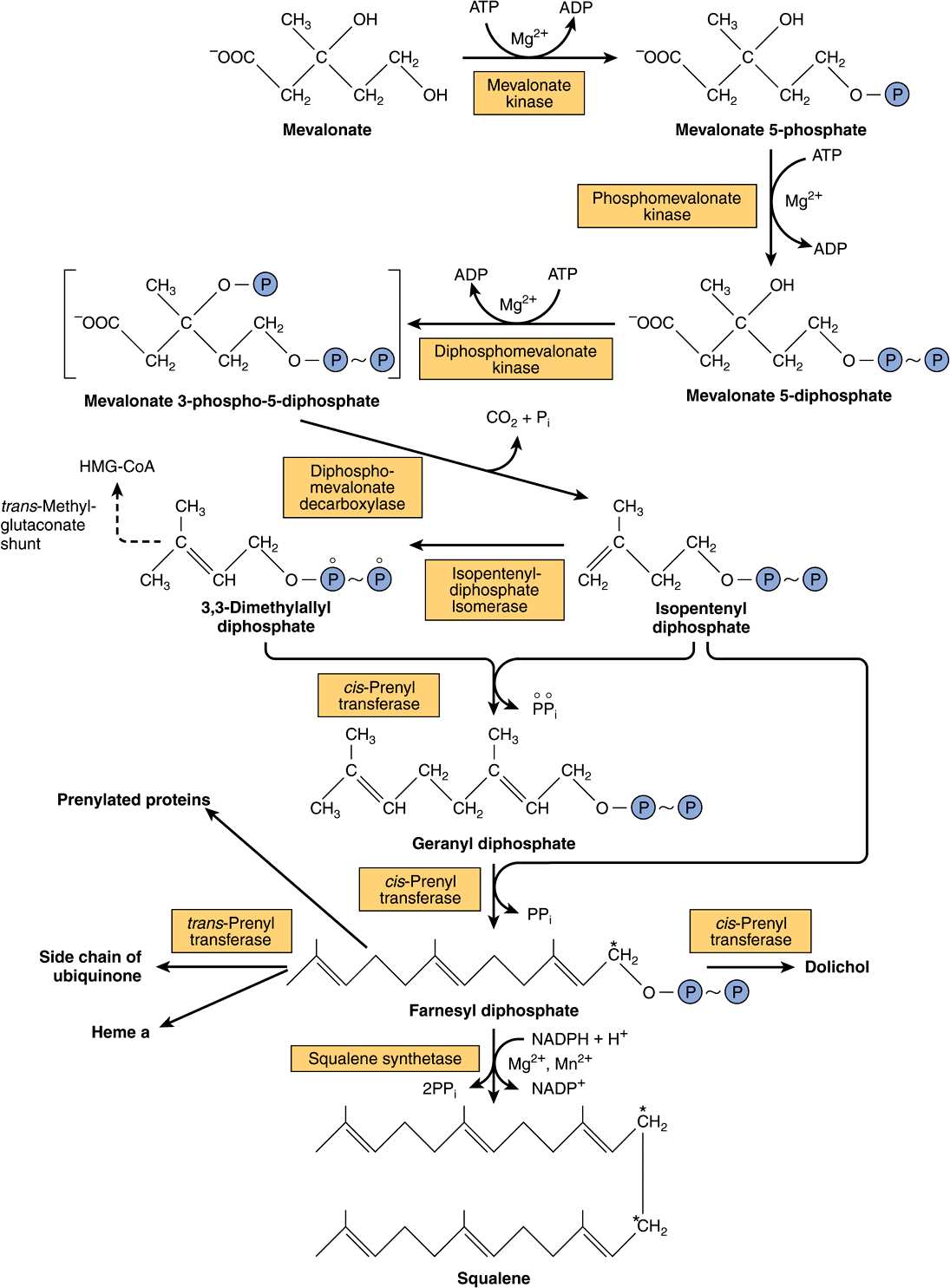
FIGURE 26–2 Biosynthesis of squalene, ubiquinone, dolichol, and other polyisoprene derivatives. (HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA.) A farnesyl residue is present in heme a of cytochrome oxidase. The carbon marked with an asterisk becomes C11 or C12 in squalene. Squalene synthetase is a microsomal enzyme; all other enzymes indicated are soluble cytosolic proteins, and some are found in peroxisomes.
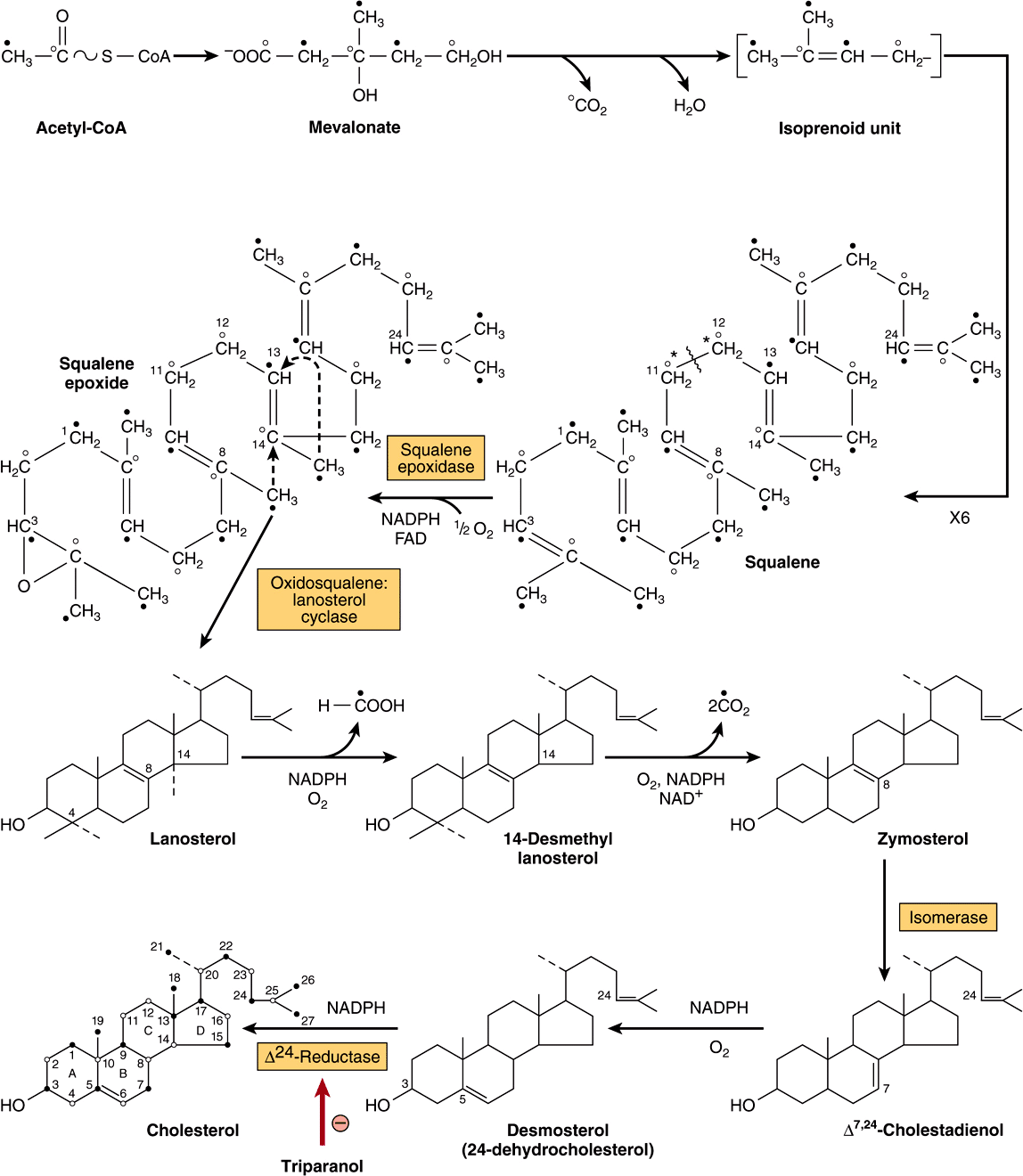
FIGURE 26–3 Biosynthesis of cholesterol. The numbered positions are those of the steroid nucleus and the open and solid circles indicate the fate of each of the carbons in the acetyl moiety of acetyl-CoA. (*Refer to labeling of squalene in Figure 26–2.)
Step 1—Biosynthesis of Mevalonate: HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) is formed by the reactions used in mitochondria to synthesize ketone bodies (Figure 22–7). However, since cholesterol synthesis is extramitochondrial, the two pathways are distinct. Initially, two molecules of acetyl-CoA condense to form acetoacetyl-CoA catalyzed by cytosolic thiolase. Acetoacetyl-CoA condenses with a further molecule of acetyl-CoA catalyzed by HMG-CoA synthase to form HMG-CoA, which is reduced to mevalonate by NADPH in a reaction catalyzed by HMG-CoA reductase. This last step is the principal regulatory step in the pathway of cholesterol synthesis and is the site of action of the most effective class of cholesterol-lowering drugs, the statins, which are HMG-CoA reductase inhibitors (Figure 26–1).
Step 2—Formation of Isoprenoid Units: Mevalonate is phosphorylated sequentially using ATP by three kinases, and after decarboxylation (Figure 26–2) the active isoprenoid unit, isopentenyl diphosphate, is formed.
Step 3—Six Isoprenoid Units Form Squalene: Isopentenyl diphosphate is isomerized by a shift of the double bond to form dimethylallyl diphosphate, and then condensed with another molecule of isopentenyl diphosphate to form the ten-carbon intermediate geranyl diphosphate (Figure 26–2). A further condensation with isopentenyl diphosphate forms farnesyl diphosphate. Two molecules of farnesyl diphosphate condense at the diphosphate end to form squalene. Initially, inorganic pyrophosphate is eliminated, forming presqualene diphosphate, which is then reduced by NADPH with elimination of a further inorganic pyrophosphate molecule.
Step 4—Formation of Lanosterol: Squalene can fold into a structure that closely resembles the steroid nucleus (Figure 26–3). Before ring closure occurs, squalene is converted to squalene 2,3-epoxide by a mixed-function oxidase in the endoplasmic reticulum, squalene epoxidase. The methyl group on C14 is transferred to C13 and that on C8 to C14 as cyclization occurs, catalyzed by oxidosqualene-lanosterol cyclase.
Step 5—Formation of Cholesterol: The formation of cholesterol from lanosterol takes place in the membranes of the endoplasmic reticulum and involves changes in the steroid nucleus and the side chain (Figure 26–3). The methyl groups on C14 and C4 are removed to form 14-desmethyl lanosterol and then zymosterol. The double bond at C8-C9 is subsequently moved to C5 -C6 in two steps, forming desmosterol. Finally, the double bond of the side chain is reduced, producing cholesterol.
Farnesyl Diphosphate Gives Rise to Dolichol & Ubiquinone
The polyisoprenoids dolichol (Figure 15–20 and Chapter 47) and ubiquinone (Figure 13–5) are formed from farnesyl diphosphate by the further addition of up to 16 (dolichol) or 3-7 (ubiquinone) isopentenyl diphosphate residues (Figure 26–2). Some GTP-binding proteins in the cell membrane are prenylated with farnesyl or geranylgeranyl (20 carbon) residues. Protein prenylation is believed to facilitate the anchoring of proteins into lipoid membranes and may also be involved in protein-protein interactions and membrane-associated protein trafficking.
CHOLESTEROL SYNTHESIS IS CONTROLLED BY REGULATION OF HMG-COA REDUCTASE
Regulation of cholesterol synthesis is exerted near the beginning of the pathway, at the HMG-CoA reductase step. The decreased synthesis of cholesterol in starving animals is accompanied by reduced activity of the enzyme. However, it is only hepatic synthesis that is inhibited by dietary cholesterol. HMG-CoA reductase in liver is inhibited by mevalonate, the immediate product of the reaction, and by cholesterol, the main product of the pathway. Cholesterol and metabolites repress transcription of the HMG-CoA reductase via activation of a sterol regulatory element-binding protein (SREBP) transcription factor. SREBPs are a family of proteins that regulate the transcription of a range of genes involved in the cellular uptake and metabolism of cholesterol and other lipids. A diurnal variation occurs both in cholesterol synthesis and reductase activity. In addition to these mechanisms regulating the rate of protein synthesis, the enzyme activity is also modulated more rapidly by post-translational modification (Figure 26–4). Insulin or thyroid hormone increases HMG-CoA reductase activity, whereas glucagon or glucocorticoids decrease it. Activity is reversibly modified by phosphorylation-dephosphorylation mechanisms, some of which may be cAMP-dependent and therefore immediately responsive to glucagon. Attempts to lower plasma cholesterol in humans by reducing the amount of cholesterol in the diet produce variable results. Generally, a decrease of 100 mg in dietary cholesterol causes a decrease of approximately 0.13 mmol/L of serum.
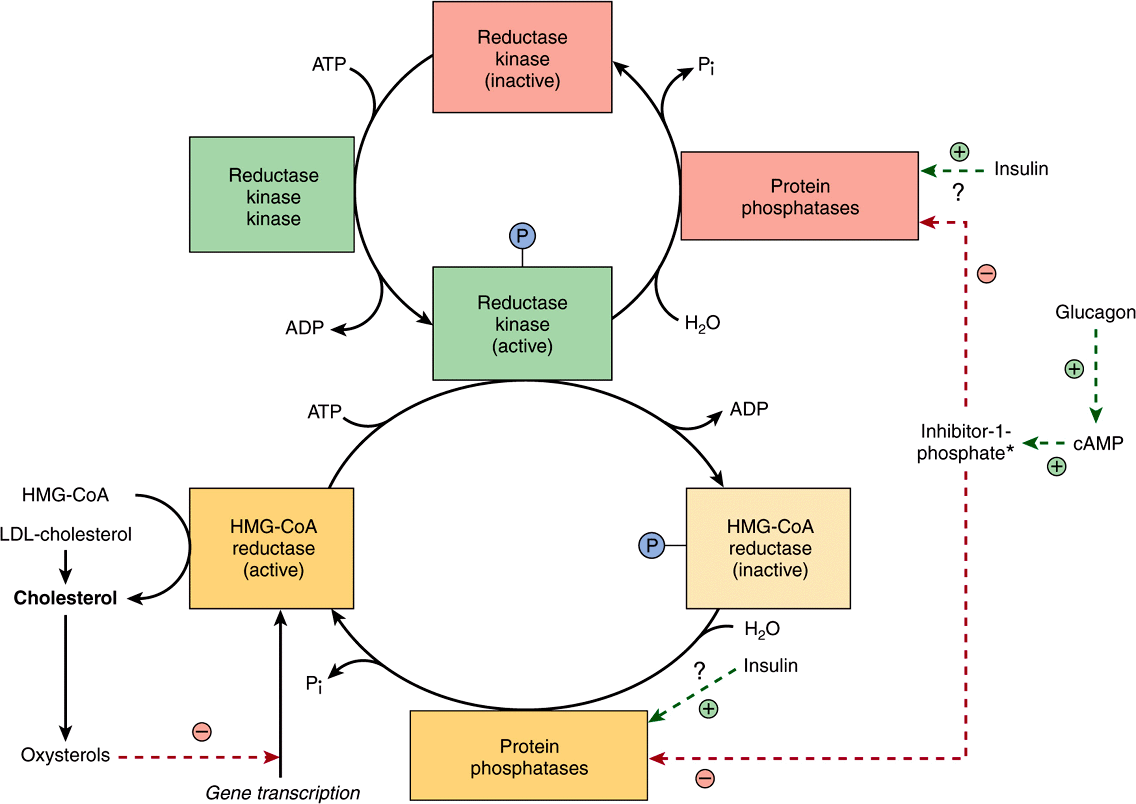
FIGURE 26–4 Possible mechanisms in the regulation of cholesterol synthesis by HMG-CoA reductase. Insulin has a dominant role compared with glucagon. (*See Figure 19–6.)
MANY FACTORS INFLUENCE THE CHOLESTEROL BALANCE IN TISSUES
In tissues, cholesterol balance is regulated as follows (Figure 26–5). An increase in cell cholesterol is caused by uptake of cholesterol-containing lipoproteins by receptors, for example, the LDL receptor or the scavenger receptor, uptake of free cholesterol from cholesterol-rich lipoproteins to the cell membrane, cholesterol synthesis, and hydrolysis of cholesteryl esters by the enzyme cholesteryl ester hydrolase. A decrease is due to efflux of cholesterol from the membrane to HDL via the ABCA1, ABCG1, or SR-B1 (Figure 25–5); esterification of cholesterol by ACAT (acyl-CoA:cholesterol acyltransferase); and utilization of cholesterol for synthesis of other steroids, such as hormones, or bile acids in the liver.
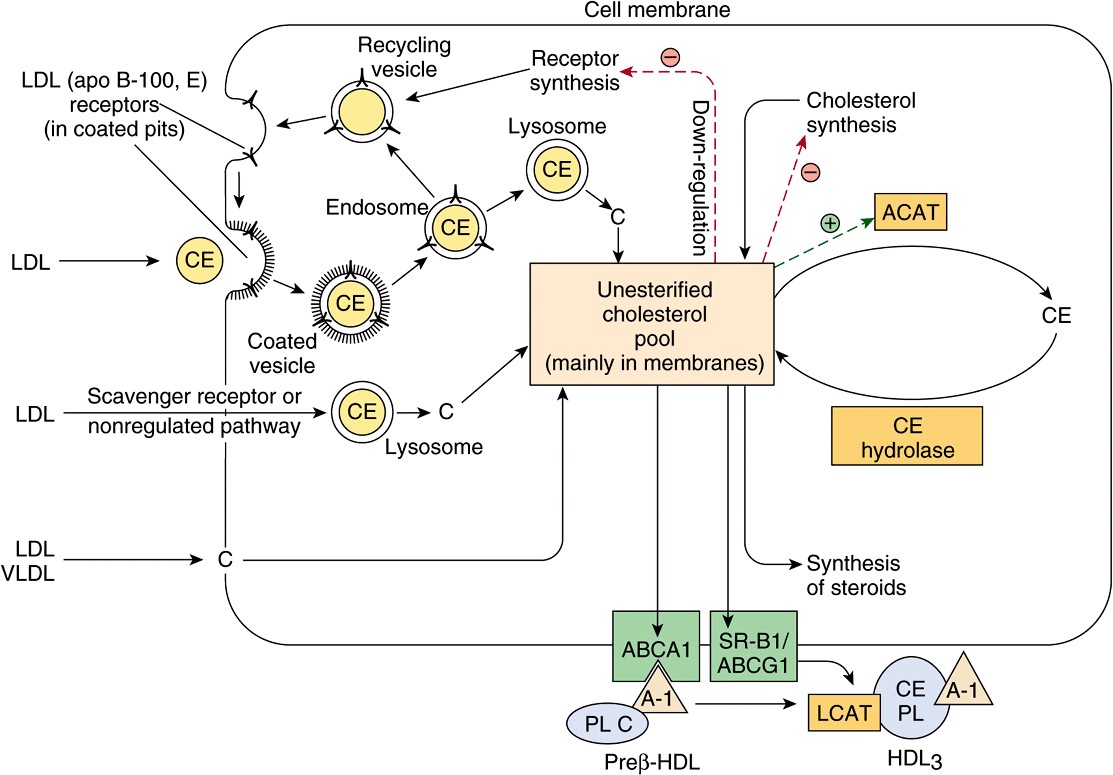
FIGURE 26–5 Factors affecting cholesterol balance at the cellular level. Reverse cholesterol transport may be mediated via the ABCA-1 transporter protein (with prepβHDL as the exogenous acceptor) or the SR-B1 or ABCG-1 (with HDL3 as the exogenous acceptor). (A-I, apolipoprotein A-I; ACAT, acyl-CoA:cholesterol acyltransferase; C, cholesterol; CE, cholesteryl ester; LCAT, lecithin:cholesterol acyltransferase; LDL, low-density lipoprotein; PL, phospholipid; VLDL, very low density lipoprotein.) LDL and HDL are not shown to scale.
The LDL Receptor Is Highly Regulated
LDL (apo B-100, E) receptors occur on the cell surface in pits that are coated on the cytosolic side of the cell membrane with a protein called clathrin. The glycoprotein receptor spans the membrane, the B-100 binding region being at the exposed amino terminal end. After binding, LDL is taken up intact by endocytosis. The apoprotein and cholesteryl ester are then hydrolyzed in the lysosomes, and cholesterol is translocated into the cell. The receptors are recycled to the cell surface. This influx of cholesterol inhibits the transcription of the genes encoding HMG-CoA synthase, HMG-CoA reductase, and other enzymes involved in cholesterol synthesis, as well as the LDL receptor itself, via the SREBP pathway, and thus coordinately suppresses cholesterol synthesis and uptake. In addition, ACAT activity is stimulated, promoting cholesterol esterification. In this way, the LDL receptor activity on the cell surface is regulated by the cholesterol requirement for membranes, steroid hormones, or bile acid synthesis (Figure 26–5).
CHOLESTEROL IS TRANSPORTED BETWEEN TISSUES IN PLASMA LIPOPROTEINS
Cholesterol is transported in plasma in lipoproteins, with the greater part in the form of cholesteryl ester (Figure 26–6), and in humans the highest proportion is found in LDL. Dietary cholesterol equilibrates with plasma cholesterol in days and with tissue cholesterol in weeks. Cholesteryl ester in the diet is hydrolyzed to cholesterol, which is then absorbed by the intestine together with dietary unesterified cholesterol and other lipids. With cholesterol synthesized in the intestines, it is then incorporated into chylomicrons (Chapter 25). Of the cholesterol absorbed, 80-90% is esterified with long-chain fatty acids in the intestinal mucosa. Ninety-five percent of the chylomicron cholesterol is delivered to the liver in chylomicron remnants, and most of the cholesterol secreted by the liver in very low density lipoprotein (VLDL) is retained during the formation of intermediate-density lipoprotein (IDL) and ultimately LDL, which is taken up by the LDL receptor in liver and extrahepatic tissues (Chapter 25).
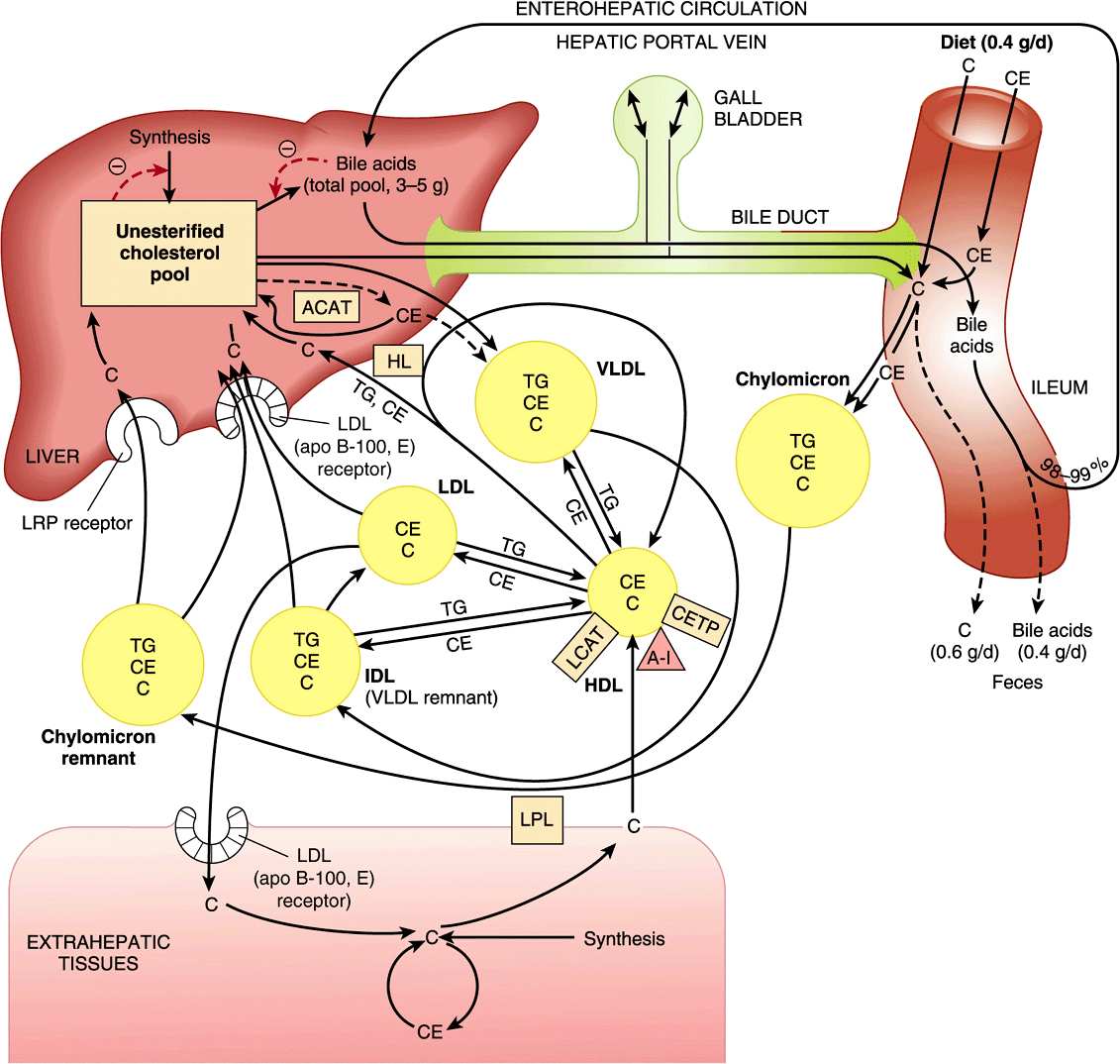
FIGURE 26–6 Transport of cholesterol between the tissues in humans. (A-I, apolipoprotein A-I; ACAT, acyl-CoA:cholesterol acyltransferase; C, unesterified cholesterol; CE, cholesteryl ester; CETP, cholesteryl ester transfer protein; HDL, high-density lipoprotein; HL, hepatic lipase; IDL, intermediate density lipoprotein; LCAT, lecithin:cholesterol acyltransferase; LDL, low-density lipoprotein; LPL, lipoprotein lipase; LRP, LDL receptor-related protein; TG, triacylglycerol; VLDL, very low density lipoprotein.)
Plasma LCAT Is Responsible for Virtually All Plasma Cholesteryl Ester in Humans
Lecithin: cholesterol acyltransferase (LCAT) activity is associated with HDL containing apo A-I. As cholesterol in HDL becomes esterified, it creates a concentration gradient and draws in cholesterol from tissues and from other lipoproteins (Figures 26-5 and 26-6), thus enabling HDL to function in reverse cholesterol transport (Figure 25–5).
Cholesteryl Ester Transfer Protein Facilitates Transfer of Cholesteryl Ester from HDL to Other Lipoproteins
Cholesteryl ester transfer protein, associated with HDL, is found in plasma of humans and many other species. It facilitates transfer of cholesteryl ester from HDL to VLDL, IDL, and LDL in exchange for triacylglycerol, relieving product inhibition of the LCAT activity in HDL. Thus, in humans, much of the cholesteryl ester formed by LCAT finds its way to the liver via VLDL remnants (IDL) or LDL (Figure 26–6). The triacylglycerol-enriched HDL2 delivers its cholesterol to the liver in the HDL cycle (Figure 25–5).
CHOLESTEROL IS EXCRETED FROM THE BODY IN THE BILE AS CHOLESTEROL OR BILE ACIDS (SALTS)
Cholesterol is excreted from the body via the bile either in the unesterified form or after conversion into bile acids in the liver. Coprostanol is the principal sterol in the feces; it is formed from cholesterol by the bacteria in the lower intestine.
Bile Acids Are Formed from Cholesterol
The primary bile acids are synthesized in the liver from cholesterol. These are cholic acid (found in the largest amount) and chenodeoxycholic acid (Figure 26–7). The 7α-hydroxylation of cholesterol is the first and principal regulatory step in the biosynthesis of bile acids and is catalyzed by cholesterol 7α-hydroxylase, a microsomal enzyme. A typical monooxygenase, it requires oxygen, NADPH, and cytochrome P450. Subsequent hydroxylation steps are also catalyzed by monooxygenases. The pathway of bile acid biosynthesis divides early into one subpathway leading to cholyl-CoA, characterized by an extra α-OH group on position 12, and another pathway leading to chenodeoxycholyl-CoA (Figure 26–7). A second pathway in mitochondria involving the 27-hydroxylation of cholesterol by sterol 27-hydroxylase as the first step is responsible for a significant proportion of the primary bile acids synthesized. The primary bile acids (Figure 26–7) enter the bile as glycine or taurine conjugates. Conjugation takes place in liver peroxisomes. In humans, the ratio of the glycine to the taurine conjugates is normally 3:1. In the alkaline bile (pH 7.6-8.4), the bile acids and their conjugates are assumed to be in a salt form—hence the term “bile salts.”
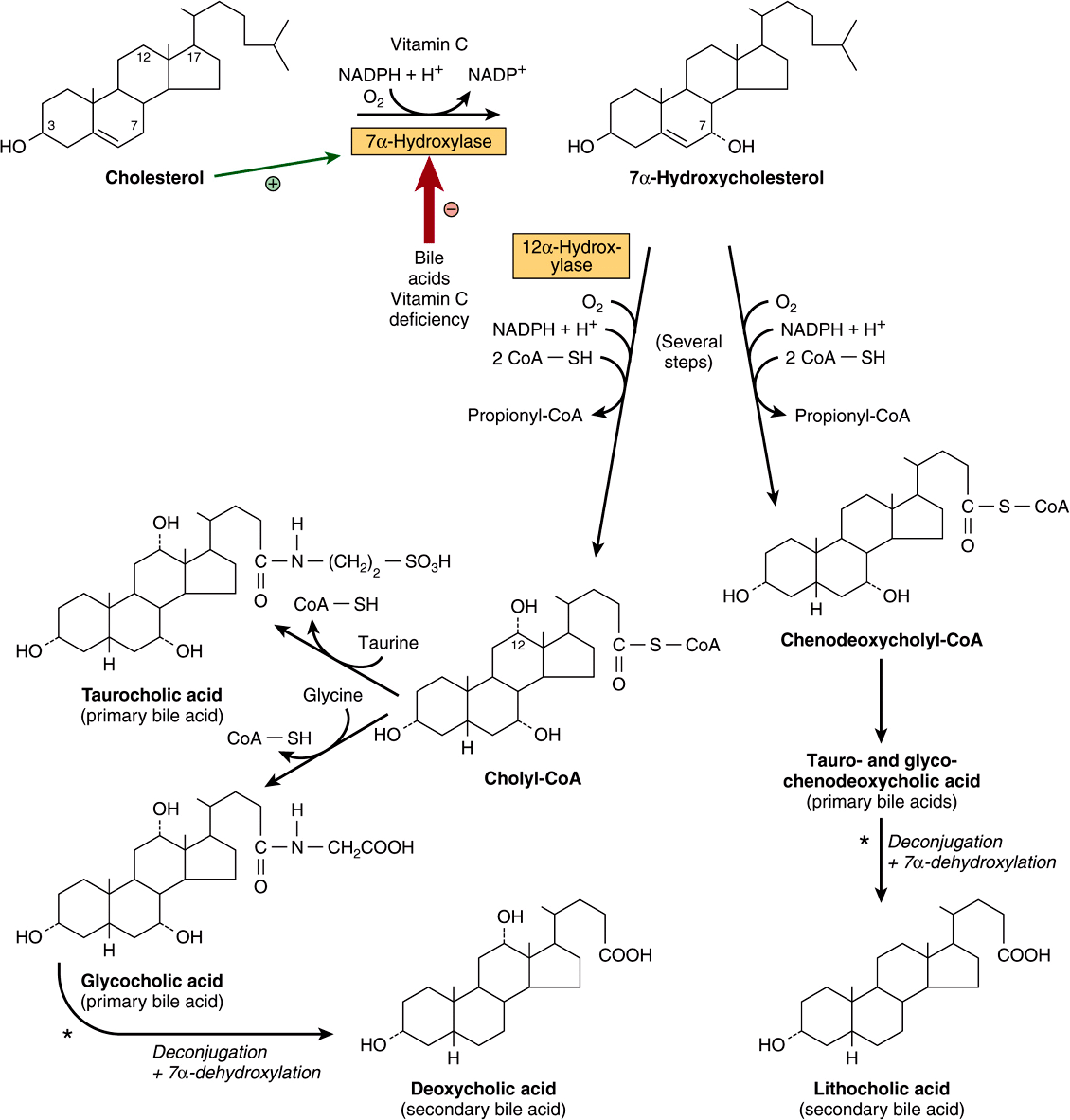
FIGURE 26–7 Biosynthesis and degradation of bile acids. A second pathway in mitochondria involves hydroxylation of cholesterol by sterol 27-hydroxylase. (*Catalyzed by microbial enzymes.)
Primary bile acids are further metabolized in the intestine by the activity of the intestinal bacteria. Thus, deconjugation and 7α-dehydroxylation occur, producing the secondary bile acids, deoxycholic acid, and lithocholic acid.
Most Bile Acids Return to the Liver in the Enterohepatic Circulation
Although products of fat digestion, including cholesterol, are absorbed in the first 100 cm of small intestine, the primary and secondary bile acids are absorbed almost exclusively in the ileum, and 98-99% is returned to the liver via the portal circulation. This is known as the enterohepatic circulation (Figure 26–6). However, lithocholic acid, because of its insolubility, is not reabsorbed to any significant extent. Only a small fraction of the bile salts escapes absorption and is therefore eliminated in the feces. Nonetheless, this represents a major pathway for the elimination of cholesterol. Each day the pool of bile acids (about 3-5 g) is cycled through the intestine six to ten times and an amount of bile acid equivalent to that lost in the feces is synthesized from cholesterol, so that a pool of bile acids of constant size is maintained. This is accomplished by a system of feedback controls.
Bile Acid Synthesis Is Regulated at the 7α-Hydroxylase Step
The principal rate-limiting step in the biosynthesis of bile acids is at the cholesterol 7α-hydroxylase reaction (Figure 26–7). The activity of the enzyme is feedback regulated via the nuclear bile acid-binding receptor, farnesoid X receptor (FXR). When the size of the bile acid pool in the enterohepatic circulation increases, FXR is activated, and transcription of the cholesterol 7α-hydroxylase gene is suppressed. Chenodeoxycholic acid is particularly important in activating FXR. Cholesterol 7α-hydroxylase activity is also enhanced by cholesterol of endogenous and dietary origin and regulated by insulin, glucagon, glucocorticoids, and thyroid hormone.
CLINICAL ASPECTS
Serum Cholesterol Is Correlated with the Incidence of Atherosclerosis & Coronary Heart Disease
While elevated plasma cholesterol levels (>5.2 mmol/L) are believed to be a major factor in promoting atherosclerosis, it is now recognized that triacylglycerols are independent risk factors. Atherosclerosis is characterized by the deposition of cholesterol and cholesteryl ester from the plasma lipoproteins into the artery wall. Diseases in which there is a prolonged elevation of levels of VLDL, IDL, chylomicron remnants, or LDL in the blood (eg, diabetes mellitus, lipid nephrosis, hypothyroidism, and other conditions of hyperlipidemia) are often accompanied by premature or more severe atherosclerosis. There is also an inverse relationship between HDL (HDL2) concentrations and coronary heart disease, making the LDL:HDL cholesterol ratio a good predictive parameter. This is consistent with the function of HDL in reverse cholesterol transport. Susceptibility to atherosclerosis varies widely among species, and humans are one of the few in which the disease can be induced by diets high in cholesterol.
Diet Can Play an Important Role in Reducing Serum Cholesterol
Hereditary factors play the most important role in determining individual serum cholesterol concentrations; however, dietary and environmental factors also play a part, and the most beneficial of these is the substitution in the diet of polyunsaturated and monounsaturated fatty acids for saturated fatty acids. Plant oils such as corn oil and sunflower seed oil contain a high proportion of polyunsaturated fatty acids, while olive oil contains a high concentration of monounsaturated fatty acids. On the other hand, butter fat, beef fat, and palm oil contain a high proportion of saturated fatty acids. Sucrose and fructose have a greater effect in raising blood lipids, particularly triacylglycerols, than do other carbohydrates.
The reason for the cholesterol-lowering effect of polyunsaturated fatty acids is still not fully understood. It is clear, however, that one of the mechanisms involved is the upregulation of LDL receptors by poly- and monounsaturated as compared with saturated fatty acids, causing an increase in the catabolic rate of LDL, the main atherogenic lipoprotein. In addition, saturated fatty acids cause the formation of smaller VLDL particles that contain relatively more cholesterol, and they are utilized by extrahepatic tissues at a slower rate than are larger particles—tendencies that may be regarded as atherogenic.
Lifestyle Affects the Serum Cholesterol Level
Additional factors considered to play a part in coronary heart disease include high blood pressure, smoking, male gender, obesity (particularly abdominal obesity), lack of exercise, and drinking soft as opposed to hard water. Factors associated with elevation of plasma FFA followed by increased output of triacylglycerol and cholesterol into the circulation in VLDL include emotional stress and coffee drinking. Premenopausal women appear to be protected against many of these deleterious factors, and this is thought to be related to the beneficial effects of estrogen. There is an association between moderate alcohol consumption and a lower incidence of coronary heart disease. This may be due to elevation of HDL concentrations resulting from increased synthesis of apo A-I and changes in activity of cholesteryl ester transfer protein. It has been claimed that red wine is particularly beneficial, perhaps because of its content of antioxidants. Regular exercise lowers plasma LDL but raises HDL. Triacylglycerol concentrations are also reduced, due most likely to increased insulin sensitivity, which enhances the expression of lipoprotein lipase.
When Diet Changes Fail, Hypolipidemic Drugs Will Reduce Serum Cholesterol & Triacylglycerol
A family of drugs known as statins have proved highly efficacious in lowering plasma cholesterol and preventing heart disease. Statins act by inhibiting HMG-CoA reductase and up-regulating the LDL receptor activity. Examples currently in use include atorvastatin, simvastatin, fluvastatin, and pravastatin. Ezetimibe reduces blood cholesterol levels by inhibiting the absorption of cholesterol by the intestine by blocking uptake via the Neimann-Pick C-like 1 protein. Other drugs used include fibrates such as clofibrate, gemfibrozil and nicotinic acid, which act mainly to lower plasma triacylglycerols by decreasing the secretion of triacylglycerol and cholesterol-containing VLDL by the liver.
Primary Disorders of the Plasma Lipoproteins (Dyslipoproteinemias) Are Inherited
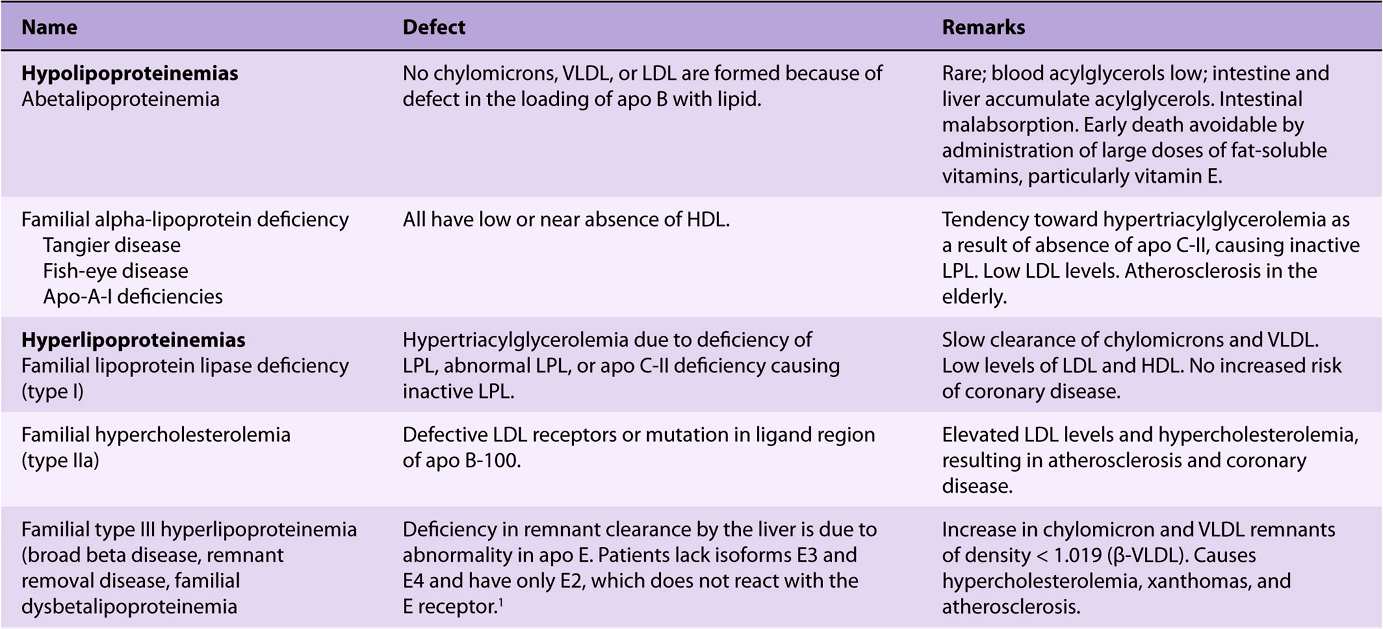
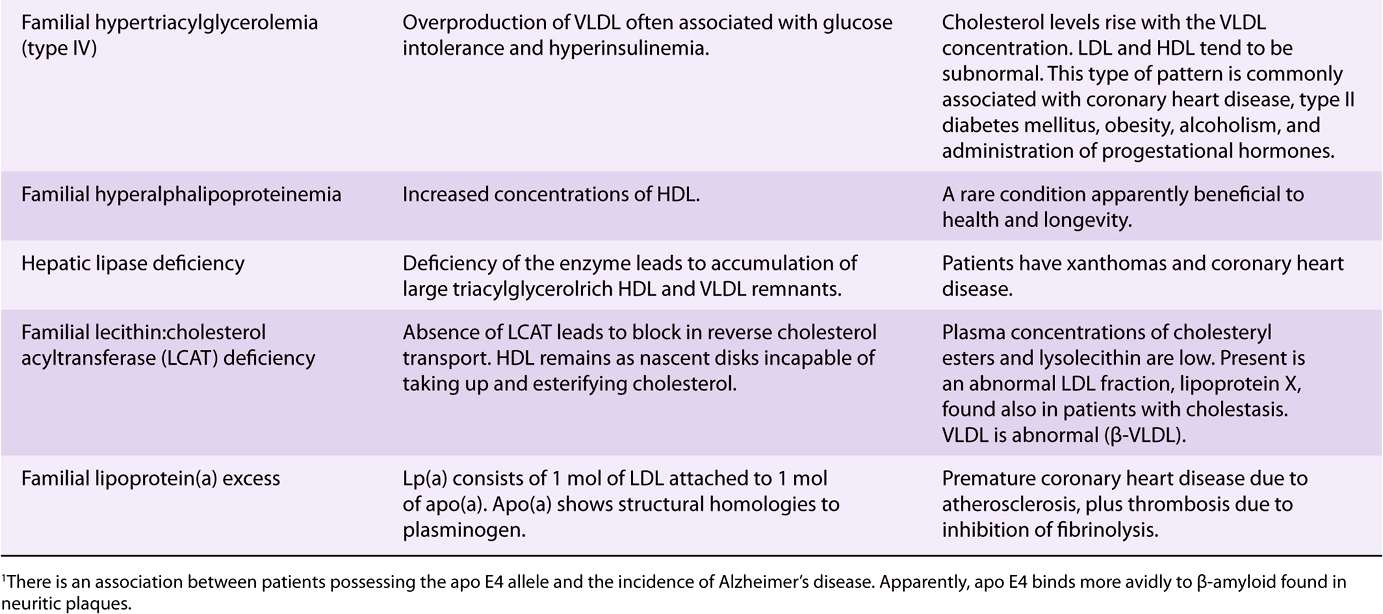
Inherited defects in lipoprotein metabolism lead to the primary condition of either hypo- or hyperlipoproteinemia (Table 26-1). In addition, diseases such as diabetes mellitus, hypothyroidism, kidney disease (nephrotic syndrome), and atherosclerosis are associated with secondary abnormal lipo-protein patterns that are very similar to one or another of the primary inherited conditions. Virtually all of the primary conditions are due to a defect at a stage in lipoprotein formation, transport, or destruction (see Figures 25-4, 26-5, and 26-6). Not all of the abnormalities are harmful.
TABLE 26–1 Primary Disorders of Plasma Lipoproteins (Dyslipoproteinemias)
SUMMARY
![]() Cholesterol is the precursor of all other steroids in the body, for example, corticosteroids, sex hormones, bile acids, and vitamin D. It also plays an important structural role in membranes and in the outer layer of lipoproteins.
Cholesterol is the precursor of all other steroids in the body, for example, corticosteroids, sex hormones, bile acids, and vitamin D. It also plays an important structural role in membranes and in the outer layer of lipoproteins.
![]() Cholesterol is synthesized in the body entirely from acetyl-CoA. Three molecules of acetyl-CoA form mevalonate via the important regulatory reaction for the pathway, catalyzed by HMG-CoA reductase. Next, a five-carbon isoprenoid unit is formed, and six of these condense to form squalene. Squalene undergoes cyclization to form the parent steroid lanosterol, which, after the loss of three methyl groups and other changes, forms cholesterol.
Cholesterol is synthesized in the body entirely from acetyl-CoA. Three molecules of acetyl-CoA form mevalonate via the important regulatory reaction for the pathway, catalyzed by HMG-CoA reductase. Next, a five-carbon isoprenoid unit is formed, and six of these condense to form squalene. Squalene undergoes cyclization to form the parent steroid lanosterol, which, after the loss of three methyl groups and other changes, forms cholesterol.
![]() Cholesterol synthesis in the liver is regulated partly by cholesterol in the diet. In tissues, cholesterol balance is maintained between the factors causing gain of cholesterol (eg, synthesis, uptake via the LDL or scavenger receptors) and the factors causing loss of cholesterol (eg, steroid synthesis, cholesteryl ester formation, excretion). The activity of the LDL receptor is modulated by cellular cholesterol levels to achieve this balance. In reverse cholesterol transport, HDL takes up cholesterol from the tissues and LCAT esterifies it and deposits it in the core of the particles. The cholesteryl ester in HDL is taken up by the liver, either directly or after transfer to VLDL, IDL, or LDL via the cholesteryl ester transfer protein.
Cholesterol synthesis in the liver is regulated partly by cholesterol in the diet. In tissues, cholesterol balance is maintained between the factors causing gain of cholesterol (eg, synthesis, uptake via the LDL or scavenger receptors) and the factors causing loss of cholesterol (eg, steroid synthesis, cholesteryl ester formation, excretion). The activity of the LDL receptor is modulated by cellular cholesterol levels to achieve this balance. In reverse cholesterol transport, HDL takes up cholesterol from the tissues and LCAT esterifies it and deposits it in the core of the particles. The cholesteryl ester in HDL is taken up by the liver, either directly or after transfer to VLDL, IDL, or LDL via the cholesteryl ester transfer protein.
![]() Excess cholesterol is excreted from the liver in the bile as cholesterol or bile salts. A large proportion of bile salts is absorbed into the portal circulation and returned to the liver as part of the enterohepatic circulation.
Excess cholesterol is excreted from the liver in the bile as cholesterol or bile salts. A large proportion of bile salts is absorbed into the portal circulation and returned to the liver as part of the enterohepatic circulation.
![]() Elevated levels of cholesterol present in VLDL, IDL, or LDL are associated with atherosclerosis, whereas high levels of HDL have a protective effect.
Elevated levels of cholesterol present in VLDL, IDL, or LDL are associated with atherosclerosis, whereas high levels of HDL have a protective effect.
![]() Inherited defects in lipoprotein metabolism lead to a primary condition of hypo- or hyperlipoproteinemia. Conditions such as diabetes mellitus, hypothyroidism, kidney disease, and atherosclerosis exhibit secondary abnormal lipoprotein patterns that resemble certain primary conditions.
Inherited defects in lipoprotein metabolism lead to a primary condition of hypo- or hyperlipoproteinemia. Conditions such as diabetes mellitus, hypothyroidism, kidney disease, and atherosclerosis exhibit secondary abnormal lipoprotein patterns that resemble certain primary conditions.
REFERENCES
Agellon LB: Metabolism and function of bile acids. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed. Vance DE, Vance JE (editors). Elsevier, 2008:423-440.
Chiang JL: Regulation of bile acid synthesis: pathways, nuclear receptors and mechanisms. J Hepatol 2004;40:539.
Denke MA: Dietary fats, fatty acids and their effects on lipoproteins. Curr Atheroscler Rep 2006;8:466.
Djoussé L, Gaziano JM: Dietary cholesterol and coronary disease risk: a systematic review. Curr Atheroscler Rep 2009;11:418.
Fernandez ML, West KL: Mechanisms by which dietary fatty acids modulate plasma lipids. J Nutr 2005;135:2075.
Jiang XC, Zhou HW: Plasma lipid transfer proteins. Curr Opin Lipidol 2006;17:302.
Liscum L: Cholesterol biosynthesis. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed. Vance DE, Vance JE (editors). Elsevier, 2008:399-422.
Ness GC, Chambers CM: Feedback and hormonal regulation of hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase: the concept of cholesterol buffering capacity. Proc Soc Exp Biol Med 2000;224:8.
Parks DJ, Blanchard SG, Bledsoe RK, et al: Bile acids: natural ligands for a nuclear orphan receptor. Science 1999;284:1365.
Perez-Sala D: Protein isoprenylation in biology and disease: general overview and perspectives from studies with genetically engineered animals. Front Biosci 2007;12:4456.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


