Inhibitors of Microbial Protein Synthesis
Drugs that inhibit protein synthesis vary considerably in terms of chemical structures and their spectrum of antimicrobial activity. Chloramphenicol, tetracyclines, and the aminoglycosides (see Chapter 45) were the first inhibitors of bacterial protein synthesis to be discovered. Because they had a broad spectrum of antibacterial activity and were thought to have low toxicities, they were overused. Many once highly susceptible bacterial species have become resistant, and most of these drugs are now used for more selected targets. Erythromycin, an older macrolide antibiotic, has a narrower spectrum of action but continues to be active against several important pathogens. Azithromycin and clarithromycin, semisynthetic macrolides, have some distinctive properties compared with erythromycin, as does clindamycin. Newer inhibitors of microbial protein synthesis, which include streptogramins, linezolid, telithromycin, and tigecycline (a tetracycline analog) have activity against certain bacteria that have developed resistance to older antibiotics.
Mechanisms of Action
Most of the antibiotics reviewed in this chapter are bacteriostatic inhibitors of protein synthesis acting at the ribosomal level (Figure 44-1). With the exception of tetracyclines, the binding sites for these antibiotics are on the 50S ribosomal subunit. Chloramphenicol inhibits transpeptidation (catalyzed by peptidyl transferase) by blocking the binding of the aminoacyl moiety of the charged transfer RNA (tRNA) molecule to the acceptor site on the ribosome-messenger (mRNA) complex. Thus, the peptide at the donor site cannot be transferred to its amino acid acceptor. Macrolides, telithromycin, and clindamycin, which share a common binding site on the 50S ribosome, also block transpeptidation. Tetracyclines bind to the 30S ribosomal subunit preventing binding of amino acid-charged tRNA to the acceptor site of the ribosome-mRNA complex.
FIGURE 44-1
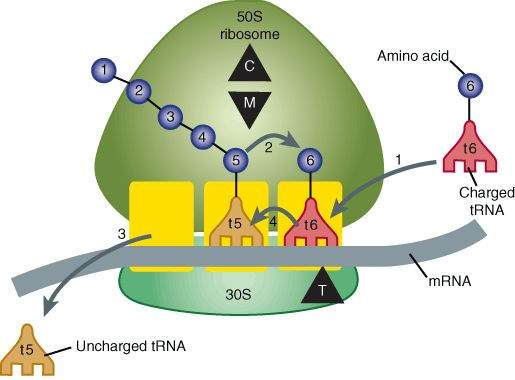
Steps in bacterial protein synthesis and targets of several antibiotics. Amino acids are shown as numbered circles. The 70S ribosomal mRNA complex is shown with its 50S and 30S subunits. In step 1, the charged tRNA unit carrying amino acid 6 binds to the acceptor site on the 70S ribosome. The peptidyl tRNA at the donor site, with amino acids 1 through 5, then binds the growing amino acid chain to amino acid 6 (transpeptidation, step 2). The uncharged tRNA left at the donor site is released (step 3), and the new 6-amino acid chain with its tRNA shifts to the peptidyl site (translocation, step 4). The antibiotic-binding sites are shown schematically as triangles. Chloramphenicol (C) and macrolides (M) bind to the 50S subunit and block transpeptidation (step 2). The tetracyclines (T) bind to the 30S subunit and prevent binding of the incoming charged tRNA unit (step 1).
(Reproduced, with permission, from Katzung BG, editor: Basic & Clinical Pharmacology, 11th ed. McGraw-Hill, 2009: Fig. 44-1.)
Streptogramins are bactericidal for most susceptible organisms. They bind to the 50S ribosomal subunit, constricting the exit channel on the ribosome through which nascent polypeptides are extruded. In addition, tRNA synthetase activity is inhibited, leading to a decrease in free tRNA within the cell. Linezolid is mainly bacteriostatic. The drug binds to a unique site on the 50S ribosome, inhibiting initiation by blocking formation of the tRNA-ribosome-mRNA ternary complex.
Selective toxicity of these protein synthesis inhibitors against microorganisms may be explained by target differences. Chloramphenicol does not bind to the 80S ribosomal RNA of mammalian cells, although it can inhibit the functions of mitochondrial ribosomes, which contain 70S ribosomal RNA. Tetracyclines have little effect on mammalian protein synthesis because an active efflux mechanism prevents their intracellular accumulation.
Chloramphenicol
Classification and Pharmacokinetics
Chloramphenicol has a simple and distinctive structure, and no other antimicrobials have been discovered in this chemical class. It is effective orally as well as parenterally and is widely distributed readily crossing the placental and blood-brain barriers. Chloramphenicol undergoes enterohepatic cycling, and a small fraction of the dose is excreted in the urine unchanged. Most of the drug is inactivated by a hepatic glucuronosyltransferase.
Antimicrobial Activity
Chloramphenicol has a wide spectrum of antimicrobial activity and is usually bacteriostatic. Some strains of Haemophilus influenzae, Neisseria meningitidis, and Bacteroides are highly susceptible, and for these organisms chloramphenicol may be bactericidal. It is not active against Chlamydia species. Resistance to chloramphenicol, which is plasmid-mediated, occurs through the formation of acetyltransferases that inactivate the drug.
Clinical Uses
Because of its toxicity, chloramphenicol has very few uses as a systemic drug. It is a backup drug for severe infections caused by Salmonella species and for the treatment of pneumococcal and meningococcal meningitis in beta-lactam-sensitive persons. Chloramphenicol is sometimes used for rickettsial diseases and for infections caused by anaerobes such as Bacteroides fragilis. The drug is commonly used as a topical antimicrobial agent
Toxicity
Gastrointestinal Disturbances
These conditions may occur from direct irritation and from superinfections, especially candidiasis.
Bone Marrow
Inhibition of red cell maturation leads to a decrease in circulating erythrocytes. This action is dose-dependent and reversible. Aplastic anemia is a rare idiosyncratic reaction (approximately 1 case in 25,000-40,000 patients treated). It is usually irreversible and may be fatal.
Gray Baby Syndrome
This syndrome occurs in infants and is characterized by decreased red blood cells, cyanosis, and cardiovascular collapse. Neonates, especially those who are premature, are deficient in hepatic glucuronosyltransferase and are sensitive to doses of chloramphenicol, which would be tolerated in older infants.
Drug Interactions
Chloramphenicol inhibits hepatic drug-metabolizing enzymes, thus increasing the elimination half-lives of drug including phenytoin, tolbutamide and warfarin.
Tetracyclines
Classification
Drugs in this class are broad-spectrum bacteriostatic antibiotics that have only minor differences in their activities against specific organisms.
Pharmacokinetics
Oral absorption is variable, especially for the older drugs, and may be impaired by foods and multivalent cations (calcium, iron, aluminum). Tetracyclines have a wide tissue distribution and cross the placental barrier. All the tetracyclines undergo enterohepatic cycling. Doxycycline is excreted mainly in feces; the other drugs are eliminated primarily in the urine. The half-lives of doxycycline and minocycline are longer than those of other tetracyclines. Tigecycline, formulated only for IV use, is eliminated in the bile and has a half-life of 30-36 h.
Antibacterial Activity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


