Fig. 51.1 The sites of action of the main classes of antibacterial drugs.
 inhibition of the synthesis of bacterial cell wall peptidoglycans or activation of enzymes that disrupt the cell wall (e.g. β-lactams),
inhibition of the synthesis of bacterial cell wall peptidoglycans or activation of enzymes that disrupt the cell wall (e.g. β-lactams), increased permeability of the bacterial cell phospholipid membrane, leading to leakage of intracellular contents (e.g. polymyxins),
increased permeability of the bacterial cell phospholipid membrane, leading to leakage of intracellular contents (e.g. polymyxins), impaired bacterial ribosome function, producing a reversible inhibition of protein synthesis (e.g. aminoglycosides, macrolides). Such drugs can show selectivity because bacterial 70S ribosomes differ structurally from the 80S ribosomes in humans,
impaired bacterial ribosome function, producing a reversible inhibition of protein synthesis (e.g. aminoglycosides, macrolides). Such drugs can show selectivity because bacterial 70S ribosomes differ structurally from the 80S ribosomes in humans,

Thirdly, antibacterials may be classified according to whether their spectrum of activity against bacteria is limited (narrow-spectrum) or extensive (broad-spectrum).
Finally, they can be classified by chemical structure. In the following text, the antimicrobial drugs are grouped according to their mechanism of action and then by their chemical structure. Cross-referencing to other methods of classification may be necessary. The drug compendium at the end of the chapter is organised to accord with the British National Formulary.
Antimicrobial resistance
When an antibacterial is ineffective against a bacterium, the organism is said to be resistant to the antibacterial drug. Resistance to antibacterial drugs can be intrinsic to the bacterium (innate resistance) or can be acquired by modification of its genetic structure (acquired resistance).
Resistance is a major problem in treating infections with bacteria, and also for many protozoa (e.g. malaria) and viruses (e.g. HIV), but is less significant in fungal infections (unless the person has immunodeficiency).
Antibacterial drug resistance
There are four general processes by which a bacterium can acquire resistance to antibacterial drugs (Fig. 51.2):
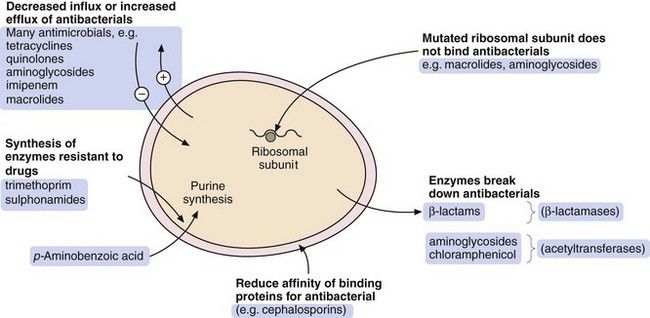
 modification of the bacterium such that it produces enzymes that inactivate the drug; examples are β-lactamase enzymes, which inactivate some penicillins, and acetylating enzymes, which can inactivate aminoglycosides,
modification of the bacterium such that it produces enzymes that inactivate the drug; examples are β-lactamase enzymes, which inactivate some penicillins, and acetylating enzymes, which can inactivate aminoglycosides, modification of the bacterium so that penetration of the drug is reduced; an example is the absence of the membrane protein D2 porin in resistant Pseudomonas aeruginosa, which prevents penetration of the β-lactam antibacterial imipenem,
modification of the bacterium so that penetration of the drug is reduced; an example is the absence of the membrane protein D2 porin in resistant Pseudomonas aeruginosa, which prevents penetration of the β-lactam antibacterial imipenem, acquisition of efflux pumps that remove the antibacterial drug from the cell faster than it can enter; an example is quinolone efflux pumps in Staphylococcus aureus,
acquisition of efflux pumps that remove the antibacterial drug from the cell faster than it can enter; an example is quinolone efflux pumps in Staphylococcus aureus, structural change in the target molecule for the antibacterial drug; examples are mutated penicillin-binding proteins in resistant enterococci that have a low affinity for binding of cephalosporins, and mutated dihydrofolate reductase that is not inhibited by trimethoprim (see Fig. 51.4, below).
structural change in the target molecule for the antibacterial drug; examples are mutated penicillin-binding proteins in resistant enterococci that have a low affinity for binding of cephalosporins, and mutated dihydrofolate reductase that is not inhibited by trimethoprim (see Fig. 51.4, below).The major mechanisms by which bacteria acquire resistance to antibacterial drugs are spontaneous mutation, conjugation, transduction and transformation.
Spontaneous mutation
In this process, a single-step genetic mutation in a bacterial population leads to resistant organisms that selectively survive and grow while sensitive bacteria are killed by an antibacterial drug. This is termed vertical evolution.
The other three mechanisms involve acquisition from other resistant organisms of genetic material that confers resistance. This is termed horizontal evolution.
Conjugation
Direct cell-to-cell contact is a way of exchanging genetic material that confers antibacterial resistance. It usually involves transfer of self-replicating circular fragments of DNA (plasmids), which can contain multiple resistance genes. A transposon (a DNA sequence that can change its relative position within the genome) may facilitate transfer of sections of DNA from one organism to another by jumping to plasmid DNA. Transfer of the plasmid occurs via a connecting structure called a pilus. The plasmid can remain outside the genome of the bacterium or can be incorporated into it, when it is more stable but less transmissible. Conjugation is by far the most important source of extrinsic DNA transfer between bacteria.
Transduction
Bacteria are susceptible to infection by viruses known as bacteriophages. During replication of the bacteriophages, the host bacterial cell’s DNA (containing resistance genes) may be replicated along with bacteriophage DNA and taken into the virus. The phage carrying the resistance genes can then infect other bacterial cells and spread resistance. This method of acquired resistance is rare.
Antibacterial drugs
The antibacterial drugs in this section are grouped by their mechanism of action and then by their chemical structure.
Drugs affecting the cell wall: β-lactam antibacterials
The drugs in this class all have a β-lactam ring which must be intact for them to be active (Fig. 51.3). The β-lactam antibacterials include penicillins, cephalosporins and cephamycins, monobactams and carbapenems. Some are susceptible to attack by bacterial enzymes (β-lactamases, also known as penicillinases) that split the β-lactam ring, but others have structural modifications that confer resistance to β-lactamase inactivation.
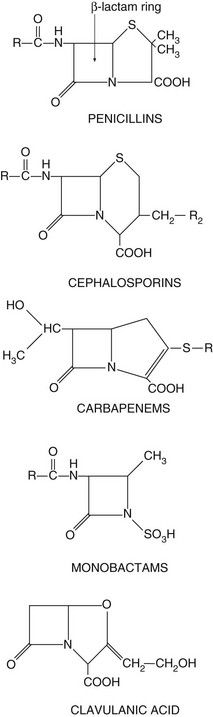
Fig. 51.3 The structural backbones of β-lactam antibacterial drugs.
Also shown is the β-lactamase inhibitor, clavulanic acid.
Mechanism of action of β-lactam antibacterials: Beta-lactam antibacterials bind to several penicillin-binding proteins in bacteria. Some of these proteins are transpeptidases that are required for crosslinking of the peptidoglycan layer of the cell wall which surrounds certain bacteria and is essential for their survival. Inhibition of transpeptidase activity by β-lactam antibacterials prevents the bacterium synthesising an intact cell wall when it divides. The transmembrane osmotic gradient then leads to swelling, rupture and death of the bacterium.
Some bacterial cells also contain enzymes that cause cell lysis when activated. The binding of β-lactam antibacterials to other specific penicillin-binding proteins within bacteria reduces the production of natural inhibitors of lysis-inducing enzymes, promoting lysis of the bacterial cell wall.
Examples
penicillins: benzylpenicillin, phenoxymethylpenicillin
aminopenicillins: amoxicillin, ampicillin, flucloxacillin
ureidopenicillins: piperacillin
amidinopenicillin: pivmecillinam
Spectrum of activity: Penicillins consist of a thiazolidine ring connected to a β-lactam ring, to which is in turn attached a side-chain (Fig. 51.3). The side-chain determines many of the antibacterial and pharmacological characteristics of particular penicillins (Table 51.1).
Table 51.1
Examples of penicillins and their properties
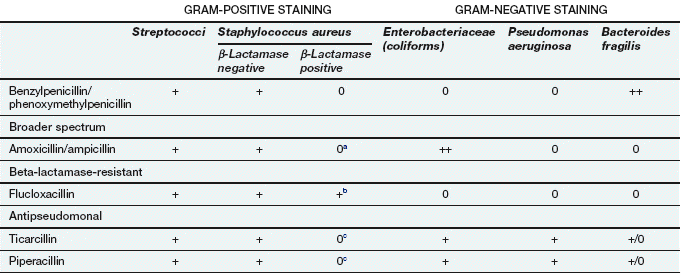
+, Active; +/0, variable activity; 0, inactive or poor activity.
aCan be used combined with a β-lactamase inhibitor, e.g. amoxicillin plus clavulanic acid (co-amoxiclav).
bResistance is increasing.
cTicarcillin only available with clavulanic acid. Piperacillin is combined with the β-lactamase inhibitor tazobactam.
Benzylpenicillin (penicillin G) and phenoxymethylpenicillin (penicillin V) are active against many aerobic Gram-positive bacteria, a more limited range of Gram-negative bacteria, for example cocci (e.g. gonococci and meningococci), and many anaerobic micro-organisms. Gram-negative bacilli are not sensitive to these drugs. Benzylpenicillin and phenoxymethylpenicillin are only effective against organisms that do not produce β-lactamases (see below).
Flucloxacillin has an acyl side chain attached to the β-lactam ring which prevents access of β-lactamase to the ring and makes the drug resistant to inactivation by the enzyme. Flucloxacillin is generally less effective than benzylpenicillin against bacteria that do not produce β-lactamase, and is usually reserved for treating β-lactamase-producing staphylococci.
Ampicillin and amoxicillin are aminopenicillins that have an extended spectrum of activity to include many Gram-negative bacilli. However, they are less effective than benzylpenicillin against Gram-positive cocci. Both drugs are inactivated by β-lactamase.
Other extended-spectrum penicillins include ureidopenicillins (e.g. piperacillin), which are active against P. aeruginosa, and amidinopenicillins (e.g. pivmecillinam), which are active mainly against Gram-negative bacteria. Carboxypenicillins (e.g. ticarcillin) are not widely used, but have activity against Pseudomonas species, Proteus species and Bacteroides fragilis.
Clavulanic acid is a potent inhibitor of several β-lactamases. It is structurally related to the β-lactam antibiotics, although it has little intrinsic antibacterial activity (Fig. 51.3). It is available in combined formulations with penicillins that are destroyed by β-lactamase, such as amoxicillin (as co-amoxiclav) or ticarcillin (Table 51.1); the combination drugs can be used to treat infections caused by some β-lactamase-producing organisms that would otherwise be resistant to the antibacterial. Tazobactam has similar properties to clavulanic acid and is used in combination with piperacillin.
Resistance: Resistance to penicillins is most often due to the production of β-lactamases which hydrolyse the β-lactam ring (Fig. 51.3). There are hundreds of β-lactamases, many of which are closely related to penicillin-binding proteins, but some are structurally different metalloenzymes. The β-lactamases produced by various organisms have widely differing spectra of activity. Some bacteria release extracellular β-lactamases, particularly S. aureus. In Gram-negative bacteria the β-lactamases are located between the inner and outer cell membranes in the periplasmic space. Extended-spectrum β-lactamases (ESBLs) are β-lactamases that also hydrolyse extended-spectrum ‘third-generation’ cephalosporins, such as cefotaxime and ceftriaxone, and monobactams such as aztreonam (see below). ESBLs are most often produced by enterobacteria. The genetic information for β-lactamase production is often encoded in a plasmid and this may be transferred to other bacteria by conjugation. By contrast, the broader-spectrum β-lactamases are often encoded by chromosomal genes.
An alternative type of penicillin resistance occurs in gonococci and in meticillin-resistant S. aureus (MRSA), which develop mutated penicillin-binding proteins that do not bind β-lactam antibacterials. Meticillin has now been discontinued, but the name MRSA is still used.
Pharmacokinetics: Only about one-third of an oral dose of benzylpenicillin (penicillin G) is absorbed; the rest is destroyed by acid in the stomach. Benzylpenicillin is therefore restricted to intramuscular or intravenous administration. The phenoxymethyl derivative (penicillin V) is more acid-stable and better absorbed from the gut; it has a similar spectrum of activity as benzylpenicillin but is generally less active. Penicillins are widely distributed through the body, although transport across the meninges is poor unless they are acutely inflamed (e.g. in meningitis). The half-lives of benzylpenicillin and phenoxymethylpenicillin, in common with most penicillins, are short (about 1 h) because they are rapidly eliminated by the kidney, mainly by active secretion at the proximal tubule. Flucloxacillin and amoxicillin are rapidly and almost completely absorbed from the gut, but ampicillin is incompletely absorbed. These drugs can also be given intramuscularly or intravenously. They are eliminated by the kidney in a similar way to benzylpenicillin. The amidinopenicillin pivmecillinam is a prodrug for oral use which is hydrolysed to mecillinam. The carboxypenicillin ticarcillin is only available in combination with clavulanic acid for intravenous use. The ureidopenicillin piperacillin is given intravenously in combination with the β-lactamase inhibitor tazobactam.
Unwanted effects: Penicillins are normally well tolerated and have a high therapeutic index.

 Allergic reactions in 1–10% of exposed individuals. Penicillins and their breakdown products bind to proteins and act as haptens, stimulating the production of antibodies that mediate the allergic response (Chs 38 and 53). Manifestations of allergy to penicillins include fever, vasculitis, serum sickness, exfoliative dermatitis, Stevens–Johnson syndrome and anaphylactic shock. Cross-allergenicity is widespread among various penicillins and to a lesser extent with cephalosporins.
Allergic reactions in 1–10% of exposed individuals. Penicillins and their breakdown products bind to proteins and act as haptens, stimulating the production of antibodies that mediate the allergic response (Chs 38 and 53). Manifestations of allergy to penicillins include fever, vasculitis, serum sickness, exfoliative dermatitis, Stevens–Johnson syndrome and anaphylactic shock. Cross-allergenicity is widespread among various penicillins and to a lesser extent with cephalosporins. Aminopenicillins (e.g. amoxicillin) frequently produce a non-allergic maculopapular rash in people with glandular fever. This does not recur if another type of penicillin is given.
Aminopenicillins (e.g. amoxicillin) frequently produce a non-allergic maculopapular rash in people with glandular fever. This does not recur if another type of penicillin is given.
 Encephalopathy with excessively high cerebrospinal fluid (CSF) concentrations of penicillin. This occurs in severe renal failure or after inadvertent intrathecal injection (which should never be given).
Encephalopathy with excessively high cerebrospinal fluid (CSF) concentrations of penicillin. This occurs in severe renal failure or after inadvertent intrathecal injection (which should never be given). Diarrhoea or Clostridium difficile-related colitis as a result of disturbance of normal colonic flora, especially with broad-spectrum penicillins.
Diarrhoea or Clostridium difficile-related colitis as a result of disturbance of normal colonic flora, especially with broad-spectrum penicillins.
Examples
first generation: cefadroxil, cefalexin
second generation: cefaclor, cefuroxime
third generation: cefotaxime, cefixime, ceftazidime, ceftriaxone
Spectrum of activity: Cephalosporins, like penicillins, have a β-lactam ring, to which is fused a dihydrothiazine ring, which makes them more resistant to hydrolysis by β-lactamases (Fig. 51.3). Cephalosporins are often classified by ‘generations’, the members within each generation sharing similar antibacterial activity. Successive generations tend to have increased activity against Gram-negative bacilli, usually at the expense of Gram-positive activity, and an increased ability to cross the blood–brain barrier (Table 51.2).
Table 51.2
Examples of β-lactams other than penicillins and their spectra of activity
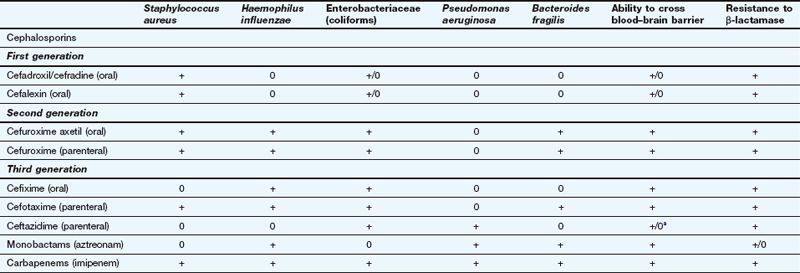
This table is a general guide to selected drugs and susceptibilities of organisms can vary widely.
Staphylococcus aureus is a Gram-positive staining organism. Other illustrative bacteria are Gram-negative staining.
aSome cephalosporins penetrate better into the CNS in the presence of inflamed meninges.
 First-generation cephalosporins (e.g. cefadroxil or cefalexin) have activity against staphylococci and most streptococci, but not enterococci.
First-generation cephalosporins (e.g. cefadroxil or cefalexin) have activity against staphylococci and most streptococci, but not enterococci. Second-generation cephalosporins (e.g. cefuroxime) have additional activity against some Gram-negative bacteria such as Haemophilus influenzae and Neisseria gonorrhoeae. They are able to penetrate the blood–brain barrier.
Second-generation cephalosporins (e.g. cefuroxime) have additional activity against some Gram-negative bacteria such as Haemophilus influenzae and Neisseria gonorrhoeae. They are able to penetrate the blood–brain barrier. Third-generation cephalosporins have improved β-lactamase stability and are able to penetrate the CSF in useful quantities. They also have greater Gram-negative activity than the other two generations. Cefixime adds Proteus and Klebsiella species to its spectrum, but it has no activity against staphylococci (Table 51.2). Ceftazidime has good activity against Pseudomonas species.
Third-generation cephalosporins have improved β-lactamase stability and are able to penetrate the CSF in useful quantities. They also have greater Gram-negative activity than the other two generations. Cefixime adds Proteus and Klebsiella species to its spectrum, but it has no activity against staphylococci (Table 51.2). Ceftazidime has good activity against Pseudomonas species.Resistance: The later generations are more resistant to β-lactamase-mediated enzymatic hydrolysis of the β-lactam ring than are the earlier generations (Table 51.2). However, ESBLs can be acquired by Escherichia coli and and other enterobacteria, which confers resistance to third-generation cephalosporins.
Pharmacokinetics: First-generation oral cephalosporins are well absorbed from the gut. Several second- and third-generation drugs, for example cefuroxime and cefotaxime, are acid-labile and must be given by a parenteral route. Cefuroxime has been formulated as a prodrug for oral use (cefuroxime axetil), which is hydrolysed to cefuroxime at first pass through the liver. Most cephalosporins are excreted primarily by the kidney and have short half-lives (less than 3 h), but cefixime is mainly eliminated by biliary excretion. Ceftriaxone has a longer half-life (6–9 h), probably as a result of extensive plasma protein binding.





Aztreonam is a β-lactam antibacterial related to the penicillins but with a single ring structure (‘monocyclic β-lactam’) (Fig. 51.3). It has little cross-allergenicity with the penicillins and has been successfully given to people with proven penicillin allergy. Its spectrum of activity is limited to Gram-negative bacteria, including P. aeruginosa, Neisseria meningitidis, N. gonorrhoeae and H. influenzae, with no activity against Gram-positive bacteria or anaerobes. Aztreonam is given intramuscularly or intravenously and is resistant to most β-lactamases. However, ESBLs can be acquired by E. coli and other enterobacteria, which confer resistance to aztreonam. Aztreonam is excreted by the kidney and has a half-life of about 2 h. Unwanted effects are similar to those of other β-lactam antibacterials.
Imipenem is a β-lactam drug that has an extremely broad spectrum of bactericidal activity. It has potent activity against Gram-positive cocci, including some β-lactamase-producing pneumococci (Table 51.2), Gram-negative bacilli, including P. aeruginosa, Neisseria suppurans and Bacteroides species, and also many anaerobic bacteria. Imipenem can penetrate the blood–brain barrier and is resistant to β-lactamases. Narrow-spectrum resistance to imipenem in P. aeruginosa occurs from a mutation that results in loss of a specific cell membrane uptake pathway.
Meropenem and ertapenem are structurally related and have broad spectra of activity against Gram-positive and Gram-negative bacteria, but ertapenem is inactive against Pseudomonas species. Imipenem, meropenem and ertapenem are given intravenously; imipenem can also be given by deep intramuscular injection. Imipenem is rapidly hydrolysed by dihydropeptidases in the kidney and so is given in combination with the dihydropeptidase inhibitor cilastatin. Meropenem and ertapenem are not inactivated by the renal dihydropeptidase and can be given alone.
These drugs are mainly excreted by the kidney and have short half-lives (1–5 h). Unwanted effects are similar to those of other β-lactam antibacterials, except for neurotoxicity with seizures, which is more common with imipenem than with other carbapenems.
Other drugs affecting the cell wall
Mechanism of action: Vancomycin and teicoplanin are high-molecular-weight glycopeptide compounds that inhibit bacterial cell wall synthesis by preventing the linking of peptidoglycan constituents (Fig. 51.1). Glycopeptides are bactericidal.
Spectrum of activity: Vancomycin and teicoplanin are active only against Gram-positive bacteria, particularly meticillin-resistant staphylococci. They do not penetrate the cell wall of Gram-negative bacteria. Both are usually reserved for treatment of serious Gram-positive bacterial infection or for bacterial endocarditis that is not responding to other treatments. Vancomycin given orally is also effective against C. difficile, which colonises the colon when the normal gut flora is disturbed by antibacterial drugs, causing diarrhoea and colitis. Metronidazole (see below) is preferred for this indication, but resistance to metronidazole is increasingly common.
Resistance: Acquired resistance to vancomycin is uncommon. In S. aureus it arises as a result of a multi-step genetic acquisition of a thickened peptidoglycan cell wall. This traps the drug and prevents it reaching its target on the cytoplasmic membrane. For other bacteria, plasmid-mediated resistance involves incorporation of D-lactate into the cell wall in place of D-alanine. This modification prevents binding of the glycopeptide.
Pharmacokinetics: Both vancomycin and teicoplanin are poorly absorbed orally and are given by intravenous infusion for systemic infection. Teicoplanin can also be given by intramuscular injection. Oral vancomycin is only used for treating C. difficile-related colitis. Both drugs are excreted by the kidney; vancomycin has a shorter half-life (5–11 h) than teicoplanin (32–176 h). Plasma concentration monitoring of vancomycin is used to minimise the risk of toxicity.
Unwanted effects: Dose adjustment based on monitoring of the trough plasma concentration of vancomycin may reduce the risk of toxic effects.






Mechanism of action: Daptomycin is a lipopeptide antibacterial with a unique mode of action. It binds to the cell wall of Gram-positive bacteria, and creates transmembrane channels that allow leakage of intracellular ions, destroying the membrane potential across the cell.
Spectrum of activity: Daptomycin does not penetrate the membrane of Gram-negative bacteria. It is bactericidal against a similar spectrum of organisms as vancomycin and is used to treat complicated skin and soft tissue infections.
Resistance: Resistance occurs when the bacterial membrane structure changes to prevent binding of the drug.
Pharmacokinetics: Daptomycin is given intravenously and eliminated unchanged by the kidneys, with a half-life of about 8 h.


Mechanism of action: Polymyxins bind to membrane phospholipids in susceptible bacteria and alter the permeability of the membrane to K+ and Na+ ions. The cell’s osmotic barrier is lost and the bacteria are killed by lysis (Fig. 51.1).
Spectrum of activity: Polymyxins have bactericidal action against Gram-negative bacteria, including Pseudomonas species, but are inactive against Gram-positive bacteria.
Pharmacokinetics: Colistimethate sodium is very poorly absorbed from the gut and is usually given by inhalation or topically to the skin. Penetration into joint spaces and CSF is poor. It is excreted unchanged by the kidney and has a half-life of 4–8 h. It is occasionally given by mouth for bowel sterilisation.



Drugs affecting bacterial DNA
Quinolones (fluoroquinolones):
Mechanism of action: Quinolones inhibit replication of bacterial DNA. They block the activity of bacterial DNA gyrase and DNA topoisomerase, the enzymes that form DNA supercoils and are essential for DNA replication and repair (Fig. 51.1). The effect is bactericidal.
Spectrum of activity: Ciprofloxacin has a broad spectrum of activity and is active against many micro-organisms resistant to penicillins, cephalosporins and aminoglycosides. Its spectrum includes Gram-positive bacteria, but with only moderate activity against Streptococcus pneumoniae and Enterococcus faecalis. It is active against most Gram-negative bacteria, including H. influenzae, P. aeruginosa, N. gonorrhoeae, and Enterobacter and Campylobacter species. Its spectrum extends to chlamydia and some mycobacteria, but not anaerobes.
Moxifloxacin has a broad spectrum of activity against Gram-positive and Gram-negative bacteria, but is inactive against P. aeruginosa. It has greater activity than ciprofloxacin against pneumococci. Norfloxacin is mainly useful for urinary tract pathogens.
Resistance: Resistance to quinolones is relatively uncommon but can be produced by a mutation that results in a DNA gyrase that is less susceptible to the drug’s action, or by increased active drug efflux from the cell (Fig. 51.2).
Pharmacokinetics: Oral absorption of ciprofloxacin is variable but adequate. Intravenous formulations of some quinolones are available, including ciprofloxacin and moxifloxacin. Ciprofloxacin is widely distributed but CSF penetration is poor unless there is meningeal inflammation. Ciprofloxacin and norfloxacin are eliminated mainly by the kidney and have short half-lives (3–4 h). Moxifloxacin is well absorbed from the gut, is metabolised in the liver and has a longer half-life (12 h).

 Central nervous system (CNS) effects: dizziness, headache, tremor, seizures (especially in those with a history of epilepsy).
Central nervous system (CNS) effects: dizziness, headache, tremor, seizures (especially in those with a history of epilepsy).
 Pain and inflammation in tendons, occasionally with tendon rupture (especially in the elderly or with concomitant use of corticosteroids).
Pain and inflammation in tendons, occasionally with tendon rupture (especially in the elderly or with concomitant use of corticosteroids). Moxifloxacin prolongs the Q–T interval on the electrocardiogram (ECG) and predisposes to ventricular arrhythmias. The risk is greater if used in combination with other proarrhythmic drugs (Ch. 8).
Moxifloxacin prolongs the Q–T interval on the electrocardiogram (ECG) and predisposes to ventricular arrhythmias. The risk is greater if used in combination with other proarrhythmic drugs (Ch. 8).
Mechanism of action: Metronidazole and tinidazole are bactericidal only after they have been converted to an intermediate transient toxic metabolite, which inhibits bacterial DNA synthesis and breaks down existing DNA. Only some anaerobes and some protozoa contain the oxidoreductase enzyme that converts these drugs to their antibacterial derivatives. The intermediate metabolite is not produced in human cells, or in aerobic bacteria. These drugs are equally active against dividing and non-dividing cells.
Spectrum of activity: Metronidazole and tinidazole are mainly active against anaerobic bacteria and protozoa, including B. fragilis, Clostridium species, Gardnerella vaginalis and Giardia lamblia. Metronidazole is an important drug for treating C. difficile-related colitis caused by broad-spectrum antimicrobial use. Metronidazole or tinidazole are important constituents of the triple or quadruple therapy utilised for the elimination of Helicobacter pylori (Ch. 33). They are also amoebicidal, with activity against Entamoeba histolytica.
Resistance: Acquired resistance is becoming more common. For example, in some countries a significant percentage of strains of H. pylori are resistant to metronidazole, as are some strains of C. difficile. Resistance can result from the development of oxidoreductases that do not act on metronidazole, or from the induction of oxidative stress mechanisms that inhibit the action of the drug. Resistance to tinidazole is less common.
Pharmacokinetics: Metronidazole is well absorbed orally and can also be given intravenously or by rectal suppositories. Metronidazole penetrates well into body fluids, including vaginal, pleural and cerebrospinal fluids, and can cross the placenta. Metronidazole and tinidazole are eliminated mainly by metabolism in the liver and have half-lives of 6–9 h and 12–14 h, respectively.



Mechanism of action: Nitrofurantoin is activated inside bacteria by reduction via the flavoprotein nitrofurantoin reductase to unstable metabolites, which disrupt ribosomal RNA, DNA and other intracellular components. It is bactericidal, especially to bacteria present in acid urine.
Spectrum of activity: Nitrofurantoin is active against most Gram-positive cocci and E. coli. Pseudomonas species are naturally resistant, as are many Proteus species. Its use is confined to infections of the lower urinary tract.
Pharmacokinetics: Nitrofurantoin is well absorbed from the gut. Its half-life in plasma is very short (<1 h) and therapeutic plasma concentrations are not achieved. It is excreted largely unchanged in the urine, giving urinary concentrations high enough to treat lower urinary tract infections, but the low tissue concentrations are inadequate for the treatment of acute pyelonephritis.



Drugs affecting bacterial protein synthesis
Mechanism of action: Macrolides interfere with bacterial protein synthesis by binding reversibly to the 50S subunit of the bacterial ribosome. This causes dissociation of the aminoacyl-transfer RNA (tRNA) from its translocation site. The action is primarily bacteriostatic (Fig. 51.1).
Spectrum of activity: Erythromycin has a similar spectrum of activity to broad-spectrum penicillins and is often used for treating individuals who are allergic to penicillin. It is effective against Gram-positive bacteria and gut anaerobes, but has poor activity against H. influenzae. It is also used for infections by Legionella, Mycoplasma, Chlamydia, Mycobacterium and Campylobacter species and for Bordetella pertussis. Although erythromycin is primarily bacteriostatic, it is bactericidal at high concentrations for some Gram-positive species, such as group A streptococci and pneumococci.
Azithromycin has less activity than erythromycin against Gram-positive bacteria, but enhanced activity against H. influenzae. Clarithromycin has slightly greater activity than erythromycin and is also used as part of the multidrug treatment of H. pylori (Ch. 33). Telithromycin is a derivative of erythromycin active against penicillin- and erythromycin-resistant S. pneumoniae.
Resistance: Bacteria become resistant to macrolides by activation of an efflux mechanism. A less common mechanism is a mutation in the gene encoding a methyltransferase that modifies the drug target site on the ribosome.
Pharmacokinetics: Erythromycin is destroyed at acid pH and is therefore given as an enteric-coated tablet or as an acid-stable ester prodrug (erythromycin ethyl succinate). It can also be administered intravenously. Clarithromycin is acid-stable and well absorbed from the gut, but undergoes first-pass metabolism in the liver. Erythromycin and clarithromycin are metabolised in the liver and have short half-lives (1–3 h). Azithromycin is poorly absorbed from the gut. It is widely distributed and released slowly from the tissues, and then excreted unchanged in the bile. It has a long half-life of about 2 days. Telithromycin is well absorbed from the gut, is metabolised in the liver and has a half-life of 10 h.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


