the rate and extent of uptake of the drug from its site of administration,


Overall, the response of an individual depends upon a combination of the effects of the drug at its site of action in the body (pharmacodynamics) and the way the body influences drug delivery to its site of action (pharmacokinetics) (Fig. 2.1). Both pharmacodynamic and pharmacokinetic aspects are subject to a number of variables, which affect the dose–response relationship. Pharmacodynamic aspects are determined by processes such as drug–receptor interaction and are specific to the class of the drug (Ch. 1). Pharmacokinetic aspects are determined by general processes, such as transfer across membranes, metabolism and renal elimination, which apply irrespective of the pharmacodynamic properties.
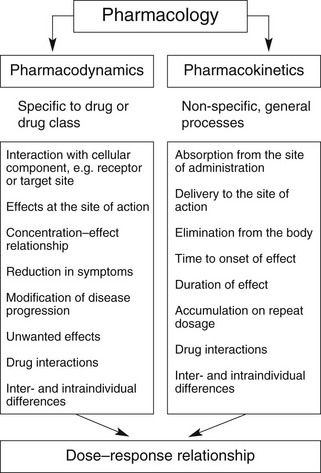
Pharmacokinetics may be divided into four basic processes, sometimes referred to collectively as ‘ADME’:

 distribution: the transfer of the drug from the general circulation into the different organs of the body,
distribution: the transfer of the drug from the general circulation into the different organs of the body,

Each of these processes can be described qualitatively in biological terms, involving biochemical and physiological processes, and also in mathematical terms, which determine many of the quantitative aspects of drug prescribing.
The biological basis of pharmacokinetics
Most drug structures bear little resemblance to normal dietary constituents such as carbohydrates, fats and proteins, and they are handled in the body by different processes. Drugs that bind to the same receptor as an endogenous ligand rarely resemble the natural ligand sufficiently closely in chemical structure to share the same carrier processes or metabolising enzymes. Consequently, the movement of drugs in the tissues is mostly by simple passive diffusion rather than by specific transporters, whereas metabolism is usually by enzymes of low substrate specificity that can handle a wide variety of drug substrates and other xenobiotics (foreign substances).
General considerations
With the exception of intravenous or intra-arterial injections, a drug must cross at least one membrane in its movement from the site of administration into the general circulation. Drugs acting at intracellular sites must also cross the cell membrane to exert an effect. The main mechanisms by which drugs can cross membranes (Fig. 2.2) are:
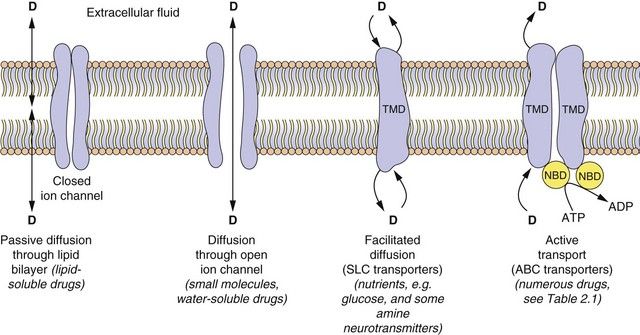
Fig. 2.2 The passage of drugs across membranes.
Molecules can cross the membrane by simple passive diffusion through the lipid bilayer or via a channel, or by facilitated diffusion, or by ATP-dependent active transport. D, drug; TMD, transmembrane domain; NBD, nucleotide-binding domain; ABC, ATP-binding cassette superfamily of transport proteins; SLC, solute carrier superfamily of transporters (see Table 2.1).




Passive diffusion: All drugs can move passively down a concentration gradient. To cross the phospholipid bilayer directly (Fig. 2.2) a drug must have a degree of lipid solubility, such as ethanol or steroids. Eventually a state of equilibrium will be reached in which equal concentrations of the diffusible form of the drug are present in solution on each side of the membrane. The rate of diffusion is directly proportional to the concentration gradient across the membrane, and to the area and permeability of the membrane, but inversely proportional to its thickness (Fick’s Law). In the laboratory, transient water-filled pores can be created in the phospholipid bilayer by applying a strong external electric field, and this process (electroporation) is used to introduce large or charged molecules, such as DNA, drugs and probes into live cells in suspension.
Passage through membrane pores or ion channels: Movement through channels occurs down a concentration gradient and is restricted to extremely small water-soluble molecules (<100 Da), such as ions. This is applicable to therapeutic ions such as lithium and also to radioactive iodine. Water itself crosses membranes rapidly via a ubiquitous family of aquaporins.
Carrier-mediated processes: Two carrier-mediated processes are of widespread importance in the transport of drugs, particularly those with low lipid solubility, across membranes.
 Active transport utilises energy (ATP) and transports drugs into or out of cells against their concentration gradient. It is performed by a family of non-specific carriers termed the ATP-binding cassette (ABC) superfamily of membrane transporters (Fig. 2.2, Table 2.1).
Active transport utilises energy (ATP) and transports drugs into or out of cells against their concentration gradient. It is performed by a family of non-specific carriers termed the ATP-binding cassette (ABC) superfamily of membrane transporters (Fig. 2.2, Table 2.1).Table 2.1
Examples of carrier molecules involved in drug transport
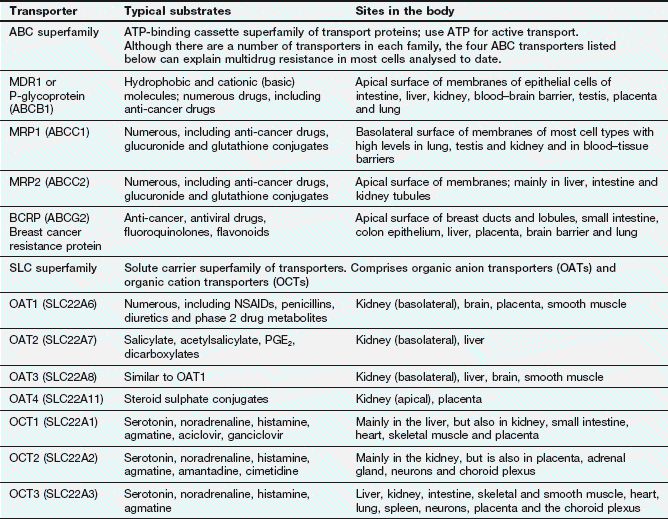
ABC, ATP-binding cassette; NSAIDs, non-steroidal anti-inflammatory drugs; PGE2, prostaglandin E2; SLC, solute carrier.
 Facilitated transport of a molecule by a carrier aids its passive movement down a concentration gradient, or uses the electrochemical gradient of a co-transported solute to transport the molecule against its own gradient; the latter is energy-dependent but does not utilise ATP. The major examples are the solute carrier (SLC) superfamily of transporters (Fig. 2.2, Table 2.1).
Facilitated transport of a molecule by a carrier aids its passive movement down a concentration gradient, or uses the electrochemical gradient of a co-transported solute to transport the molecule against its own gradient; the latter is energy-dependent but does not utilise ATP. The major examples are the solute carrier (SLC) superfamily of transporters (Fig. 2.2, Table 2.1).In humans, the ABC active-transporter superfamily contains 49 members organised into seven subfamilies (A–G) based on their relative sequence homology. Interest in this area has expanded rapidly since the discovery of P-glycoprotein (P-gp), also known as multidrug resistance 1 (MDR1) or ABCB1 transporter. P-gp transports a wide range of drug substrates, including anti-cancer drugs, steroids and immunosuppressive agents, from the cytoplasm to the extracellular side of the cell membrane, and therefore acts as an efflux transporter. Verapamil increases the concentrations of anti-cancer drugs at their intracellular sites of action by inhibiting P-gp (Ch. 52). ABCB transporter proteins contain two hydrophobic transmembrane domains, which consist of different numbers of membrane-spanning α-helices (12 in P-gp), and two hydrophilic nucleotide (ATP)-binding domains, which bind and hydrolyse intracellular ATP. The transporter is on the apical surface and acts as an efflux pump that transports substrates from the cell into the interstitial fluid, plasma, bile, urine or gut lumen. Examples of other ABC transporters are given in Table 2.1.
The SLC superfamily comprises over 400 types of organic anion transporters (OATs) and organic cation transporters (OCTs) (Table 2.1). OAT1 to OAT4 are present in various tissues; OAT1 is the classic organic anion transporter in the kidney, which secretes urate and penicillins and is blocked by probenecid (Ch. 33). Organic cation transporters (OCT1, OCT2 and OCT3) effect facilitated diffusion and can transport cations in both directions across the membrane. Substrates common to all three OCT transporters are serotonin (5-hydroxytryptamine, 5-HT), noradrenaline, histamine and agmatine; although some drugs are substrates for the transporters (Table 2.1), many basic drugs act as inhibitors of the transporters.
Pinocytosis: This can be regarded as a form of carrier-mediated entry into the cell cytoplasm. Pinocytosis is normally concerned with the uptake of endogenous macromolecules and may be involved in the uptake of recombinant therapeutic proteins; drugs can also be incorporated into a lipid vesicle or liposome for pinocytotic uptake (e.g. amphotericin and doxorubicin; Ch. 51).
Drug ionisation and membrane diffusion
Ionisation is a fundamental property of most drugs that are either weak acids, such as aspirin, or weak bases, such as propranolol. The presence of an ionisable group(s) is essential for the mechanism of action of most drugs, because ionic forces represent a key part of ligand–receptor interactions (Ch. 1). The extent of ionisation may also influence the extent of absorption of a drug, its distribution into organs such as the brain or adipose tissue and the mechanism and route of its elimination from the body.
Drugs with ionisable groups exist in equilibrium between charged (ionised) and uncharged (un-ionised) forms (Fig. 2.3). The extent of ionisation of a drug depends on the strength of the ionisable group and the pH of the solution. The extent of ionisation is given by the acid dissociation constant, Ka.
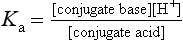
The term conjugate acid refers to a form of the drug able to release a proton, such as:
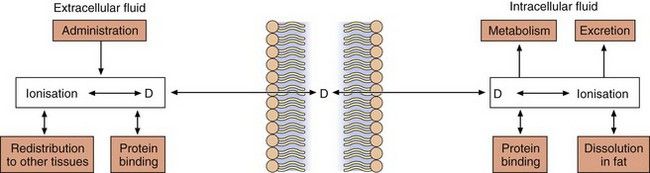
Fig. 2.3 The effect of pH on drug ionisation.
Acidic conditions (low pH, high H+ concentrations) push the equilibrium of acidic drugs towards their un-ionised (protonated) form, and basic drugs towards their ionised form. Basic conditions (high pH) have the opposite effect.


The conjugate base is the corresponding equilibrium form of the drug that has lost a proton, such as:


The value of Ka is normally much less than 1, so it is easier to compare compounds using the negative logarithm of Ka, which is called pKa. For example, a Ka of 10−5 becomes pKa 5, and a Ka of 10−10 becomes pKa 10. Based on the equation above, a strong acid (such as an  SO3H functional group) that readily donates its H+ ion will have a relatively high Ka value (e.g. 10−1 or 10−2) and hence a low pKa (i.e. 1 or 2), whereas weakly acidic groups, which donate their H+ ion less readily, have a pKa of 4–5. Conversely, for basic functional groups, the stronger the base, the greater will be its ability to retain the H+, resulting in low Ka and high pKa values. Thus, strongly basic groups have a pKa of 10–11, while weakly basic groups have a pKa of 7–8.
SO3H functional group) that readily donates its H+ ion will have a relatively high Ka value (e.g. 10−1 or 10−2) and hence a low pKa (i.e. 1 or 2), whereas weakly acidic groups, which donate their H+ ion less readily, have a pKa of 4–5. Conversely, for basic functional groups, the stronger the base, the greater will be its ability to retain the H+, resulting in low Ka and high pKa values. Thus, strongly basic groups have a pKa of 10–11, while weakly basic groups have a pKa of 7–8.
Drugs are 50% ionised when the pH of the solution equals the pKa of the drug. Acidic drugs (low pKa values) are least ionised in acidic solution (low pH) and most ionised in alkaline solutions (high pH). Basic drugs (high pKa values) are least ionised in alkaline solutions (high pH), and most ionised in acid solutions (low pH). In either case, the ionised form of the molecule can generally be regarded as the water-soluble form and the un-ionised form as the lipid-soluble form. The ease with which a drug can diffuse across a lipid bilayer is determined by the lipid solubility of its un-ionised form (Fig. 2.4).
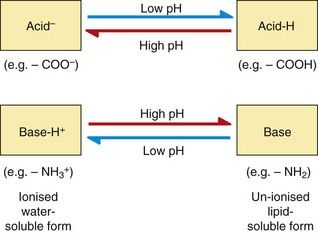
Fig. 2.4 Passive diffusion and the factors that affect drug concentrations in equilibrium between un-ionised and ionised forms.
In this case, the pH is assumed to be the same on each side of the membrane; see Fig. 2.5 for drug partitioning when there is a pH gradient across the membrane.
The pH of body fluids is controlled by the buffering capacity of the ionic groups present in endogenous molecules such as phosphate ions and proteins. When the fluids on each side of a membrane have the same pH value there will be equal concentrations of both the diffusible (un-ionised) form and the non-diffusible (ionised) form of the drug on each side of the membrane at equilibrium (Fig. 2.4).
When the fluids on each side of a membrane are at different pH values, the concentrations of the un-ionised form on each side of the membrane at equilibrium will remain equal as it can diffuse reversibly across the membrane, but the concentrations of the ionised drug will be determined by the pH of the solution. This results in pH-dependent differences in total drug concentration on each side of a membrane (pH trapping or partitioning), with the total drug concentration being higher on the side of the membrane on which it is most ionised. This is exemplified by the pH difference between urine (pH 5–7) and plasma (pH 7.4), which can influence renal elimination of drugs (Fig. 2.5). The relatively low pH of the urine forces an acidic drug to become predominantly un-ionised, allowing its reabsorption into the plasma, while the higher pH in plasma (7.4) converts the drug to the ionised form, preventing it diffusing back and trapping (partitioning) it in the plasma. The opposite situation prevails with basic drugs, which are enabled to diffuse from the plasma into urine, where they become trapped.
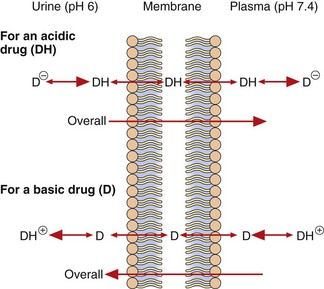
Fig. 2.5 Partitioning of acidic and basic drugs across a pH gradient.
Only the un-ionised forms (DH and D) are able to diffuse across the membrane. In urine (pH 6), the un-ionised acidic drug (DH) can be readily reabsorbed into the plasma, where its ionised form (D−) becomes concentrated, while the ionised basic drug (D+) is trapped within the urine. Alkalinising the urine would reduce reabsorption of the acid drug and enhance that of the basic drug.
After drug overdose, when the aim is to enhance drug elimination, alkalinisation of the urine can be used to reduce reabsorption of acidic drugs, leading to their faster elimination in the urine. Acidification of the urine can enhance ionisation and renal elimination of basic drugs, such as dexamfetamine.
The pH difference between gastric contents (pH 1–2) and plasma (pH 7.4) affects the absorption of many oral drugs. The acidity of stomach contents means that an acidic drug is present largely in its un-ionised (protonated) form, allowing it to pass into plasma where its ionised form becomes partitioned. In contrast, basic drugs are highly ionised in the stomach and absorption is negligible until the stomach empties and the drug can be absorbed from the more alkaline lumen of the duodenum (pH ~8).
Drugs that are fixed in their ionised form at all pH values, such as the quaternary amine compound suxamethonium (Ch. 27), cross cell membranes extremely slowly or not at all; they are given by injection (because of lack of absorption from the gastrointestinal tract) and have limited effects on the brain (because of lack of entry).
Absorption
Absorption is the process of transfer of the drug from the site of administration into the general or systemic circulation.
Absorption from the gut
The easiest and most convenient route of administration of medicines is orally by tablets, capsules or syrups. The large surface area of the small intestine combined with its high blood flow can give rapid and complete absorption of orally administered drugs. However, this route presents a number of obstacles for a drug before it reaches the systemic circulation.
Drug structure
Drug structure is a major determinant of absorption. Drugs need to be lipid-soluble to be absorbed from the gut. Highly polar acids and bases tend to be absorbed only slowly and incompletely, with much of the unabsorbed dose being voided in the faeces. High polarity may, however, be useful for delivery of the drug to a site of action in the lower bowel (see Ch. 34). The structures of some drugs can make them unstable either at the low pH of the stomach (e.g. benzylpenicillin) or in the presence of digestive enzymes (e.g. insulin). Such compounds have to be given by injection, but administration by other routes may be possible (e.g. inhalation for insulin).
Drugs that are weak acids or bases undergo pH partitioning between the gut lumen and mucosal cells. Acidic drugs will be least ionised in the stomach lumen and most absorption would be expected at this site, but absorption in the stomach is limited by its relatively low surface area (compared to the small intestine) and the presence of a zone of neutral pH on the immediate surface of the gastric mucosal cells (the mucosal bicarbonate layer; see Ch. 33). In consequence, the bulk of the absorption of drugs, even weak acids such as aspirin, occurs in the small intestine.
Drug formulation
A drug cannot be absorbed when it is taken in a tablet or capsule until the vehicle disintegrates and the drug is dissolved in the gastrointestinal contents to form a molecular solution. Most tablets disintegrate and dissolve quickly and completely and the whole dose rapidly becomes available for absorption. However, some formulations are designed to disintegrate slowly so that the rate of release and dissolution of drug from the formulation determines the rate of absorption. In modified-release (i.e. slow-release) formulations the drug is either incorporated into a complex matrix from which it diffuses slowly, or in a crystallised form that dissolves slowly. Dissolution of a tablet in the stomach can also be prevented by coating it in an acid-insoluble layer, producing enteric-coated formulations. This is useful for drugs such as omeprazole (Ch. 33), which is unstable in an acid environment, and allows delivery of intact drug to the duodenum.
Gastric emptying
The rate of gastric emptying determines how soon a drug taken orally is delivered to the small intestine, the major site of absorption. Delay between oral drug ingestion and the drug being detected in the circulation is usually caused by delayed gastric emptying. Drugs that slow gastric emptying, for example antimuscarinics, can delay the absorption of other drugs taken at the same time.
Food has complex effects on drug absorption; it slows gastric emptying and delays drug absorption, and it can also bind drugs and reduce the total amount of drug absorbed.
First-pass metabolism
Metabolism of drugs can occur before and during their absorption, and this can limit the amount of parent compound that reaches the general circulation. Drugs taken orally have to pass four major metabolic barriers before they reach the general circulation. If there is extensive metabolism of a drug at one or more of the sites below, only a fraction of the original oral dose reaches the general circulation as the parent compound. This process is known as first-pass metabolism because it occurs at the first passage through the organ.
Intestinal lumen: The intestinal lumen contains digestive enzymes secreted by the mucosal cells and pancreas that are able to split amide, ester and glycosidic bonds. Intestinal proteases prevent the oral administration of peptide drugs, such as insulin and other products of molecular biological approaches to drug development. In addition, the lower bowel contains large numbers of aerobic and anaerobic bacteria that are capable of performing a range of metabolic reactions on drugs, especially hydrolysis and reduction.
Intestinal wall: The walls of the upper intestine are rich in cellular enzymes such as monoamine oxidase (MAO), aromatic L-amino acid decarboxylase, cytochrome P450 isoenzymes (e.g. CYP3A4) and the enzymes responsible for phase 2 conjugation reactions described below. In addition, the luminal membrane of the intestinal cells (enterocytes) contains efflux transporters such as P-gp (see above), which may limit the absorption of a drug by transporting it back into the intestinal lumen. Drug molecules that enter the enterocyte may thus undergo three possible fates; that is, diffusion into the hepatic portal circulation, metabolism within the cell or transport back into the gut lumen (by P-gp). The substrate specificities of CYP3A4 and P-gp overlap and for common substrates their combined actions can prevent most of the oral dose of some drugs reaching the hepatic portal circulation.
Liver: Blood from the intestine is delivered by the splanchnic circulation directly to the liver, which is the major site of drug metabolism in the body. Hepatic first-pass metabolism can be avoided by administering the drug to a region of the gut from which the blood does not drain into the hepatic portal vein, for example the buccal cavity or rectum; a good example of this is the buccal administration of glyceryl trinitrate (Ch. 5).
Absorption from other routes
Percutaneous (transcutaneous) administration
The human epidermis (especially the stratum corneum) is an effective permeability barrier to water loss and to the transfer of water-soluble compounds. Although lipid-soluble drugs are able to cross this barrier, the rate and extent of entry are very limited. In consequence, this route is only effective for use with potent, non-irritant drugs, such as glyceryl trinitrate (Ch. 5) or fentanyl (Ch. 19), or to produce a local effect. The slow and continued absorption from dermal administration (e.g. via adhesive patches) can be used to produce low, but relatively constant, blood concentrations of some drugs; for example nicotine-replacement therapy (Ch. 54).
Intradermal and subcutaneous injection
Intradermal or subcutaneous injection avoids the barrier presented by the stratum corneum and entry into the general circulation is limited mainly by the rate of blood flow to the site of injection. However, these sites only allow the administration of small volumes of drug and tend to be used mostly for local effects, such as local anaesthesia, or to deliberately limit the rate of drug absorption, for example insulin (Ch. 40).
Intramuscular injection
The rate of absorption from an intramuscular injection depends on two variables: the local blood flow and the water solubility of the drug. An increase in either of these factors enhances the rate of removal from the injection site. Absorption of drugs from the injection site can be prolonged intentionally either by incorporation of the drug into a lipophilic vehicle, such as flupentixol decanoate (Ch. 21) or by formation of a sparingly soluble salt, such as benzathine benzylpenicillin (Ch. 51), creating a depot formulation from which the drug is released over days or weeks.
Intranasal administration
The nasal mucosa provides a good surface area for absorption, combined with low levels of proteases and drug-metabolising enzymes compared with the gastrointestinal tract. In consequence, intranasal administration is used for the administration of some drugs, such as triptan drugs for migraine (Ch. 26), and desmopressin (Ch. 43), as well as for drugs designed to produce local effects, such as nasal decongestants and topical corticosteroids (Ch. 39).
Inhalation
Although the lungs possess the characteristics of a good site for drug absorption (a large surface area and extensive blood flow), inhalation is rarely used to produce systemic effects. The principal reasons for this are the difficulty of delivering non-volatile drugs to the alveoli and the potential for local toxicity to alveolar membranes. Therefore, drug administration by inhalation is largely restricted to:

 locally acting drugs, such as bronchodilators and corticosteroids used in the treatment of airway disease such as asthma and chronic obstructive pulmonary disease (Ch. 12).
locally acting drugs, such as bronchodilators and corticosteroids used in the treatment of airway disease such as asthma and chronic obstructive pulmonary disease (Ch. 12).Drugs in the latter group are not volatile and have to be given as either aerosols containing droplets of dissolved drug or fine particles of the solid drug (dry powder) (see Ch. 12). Particles greater than 10 µm in diameter settle out in the upper airways, which are poor sites for absorption, and the drug then passes back up the airways via ciliary motion and is eventually swallowed. It was estimated that only 5–10% of an inhaled dose is absorbed from the airways, although the percentage may be higher with modern inhaler devices delivering particle sizes closer to the optimum for airways deposition (2–5 µm). Particles less than 1 µm in diameter are not deposited in the airways and are exhaled.
Distribution
Distribution is the process by which the drug is transferred reversibly from the general circulation into the tissues as the concentrations in blood increase, and from the tissues into blood when the blood concentrations decrease. For most drugs this occurs by passive diffusion of the un-ionised form across cell membranes (Fig. 2.2) until equilibrium is reached (Fig. 2.4). At equilibrium, any process that removes the drug from one side of the membrane results in movement of drug across the membrane to re-establish the equilibrium (Fig. 2.4).
After an intravenous injection there is a high initial plasma concentration and the drug may rapidly enter well-perfused tissues such as the brain, liver and lungs (Table 2.2). This may be so rapid that these tissues may be assumed to equilibrate instantaneously with plasma and represent part of the ‘central’ compartment (see below). However, the drug will continue to enter poorly perfused tissues, and this will lower the plasma concentration. The high concentrations in the rapidly perfused tissues then decrease in parallel with the decreasing plasma concentration, resulting in a transfer of drug back into the plasma (Fig. 2.6). This redistribution is important for terminating the action of some drugs given as a rapid intravenous injection or bolus. For example, intravenous thiopental produces rapid anaesthesia, but effects in the brain are short-lived because continued uptake into muscle lowers the concentrations in the blood and therefore in the brain (Fig. 2.6; see also Fig. 17.2).
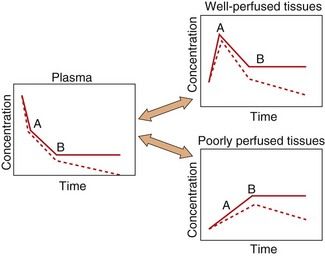
Fig. 2.6 A simplified scheme for the redistribution of drugs between tissues.
The initial decrease in plasma concentrations results from uptake into well-perfused tissues, which essentially reaches equilibrium at point A. Between points A and B, the drug continues to enter poorly perfused tissues, resulting in a decrease in the concentrations in both plasma and well-perfused tissues. At point B all tissues are in equilibrium. The additional presence of an elimination process would produce a decrease from point B (shown as a dashed line), which would be parallel in all tissues.
The processes of elimination (such as metabolism and excretion) are of major importance and are discussed in detail below. Elimination results in a net transfer of drug from other tissues via the circulation to the organ(s) of elimination (see dashed lines in Fig. 2.6).
Reversible protein binding
Many drugs show an affinity for sites on non-receptor proteins, resulting in reversible binding:
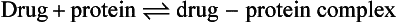
Such binding occurs with plasma proteins, most commonly albumin, which binds many acidic drugs, and α1-acid glycoprotein, which binds many basic or neutral drugs (Table 2.3). Drugs may also bind reversibly with intracellular proteins (Fig. 2.4). The drug–protein binding interaction resembles the drug–receptor interaction since it is rapid, reversible and saturable, and different ligands can compete for the same site. It does not result in a pharmacological effect but lowers the free concentration of drug available to act at receptors; the amounts of drug remaining available may be only a minute fraction of the total body load. Proteins such as albumin can therefore act as depots, rapidly releasing the bound drug when the free drug is distributed to other compartments or eliminated.
Table 2.3
Examples of drugs that undergo extensive binding to plasma protein
| Bound to albumin | Bound to α1-acid glycoprotein |
| Digitoxin | Chlorpromazine |
| Furosemide | Propranolol |
| Ibuprofen | Quinidine |
| Indometacin | Tricyclic antidepressants |
| Phenytoin | Lidocaine |
| Salicylates | |
| Sulphonamides | |
| Thiazides | |
| Tolbutamide | |
| Warfarin |
Competition for binding to proteins in plasma or inside cells can occur between different drugs (drug interaction; see Ch. 56), and also between drugs and endogenous ligands. A highly protein-bound drug such as aspirin can displace another drug such as warfarin from its binding sites on plasma proteins; the increase in unbound drug concentration can increase the biological activity of the displaced drug. An example of interaction with an endogenous ligand is the displacement of bilirubin from albumin-binding sites by sulphanilamide drugs, causing a potentially dangerous increase in the bilirubin concentration in plasma, leading to kernicterus.
Irreversible protein binding
Certain drugs, because of chemical reactivity of the parent compound or a metabolite, undergo covalent binding to plasma or tissue components such as proteins or nucleic acids. When the binding is irreversible, for example the interaction of some cytotoxic drugs with DNA, this can be considered as equivalent to elimination, because the parent drug cannot re-enter the circulation. In contrast, some covalent binding may be slowly reversible, such as the formation of disulphide bridges by captopril with its target ACE and with plasma proteins (Ch. 6); the covalently bound drug will not dissociate rapidly in response to a decrease in the concentration of unbound drug, and such binding represents a slowly equilibrating reservoir of drug.
Distribution to specific organs
Two systems require more detailed consideration of drug distribution: the brain, because of the difficulty of drug entry, and the fetus, because of the potential for drug toxicity.
Brain
Lipid-soluble drugs readily pass from the blood into the brain, and for such drugs the brain represents a typical well-perfused tissue (Table 2.2). In contrast, the entry of water-soluble drugs into the brain is much slower than into other well-perfused tissues, giving rise to the concept of a blood–brain barrier. The functional basis of the barrier to water-soluble drugs (Fig. 2.7) is reduced permeability of brain capillaries owing to:
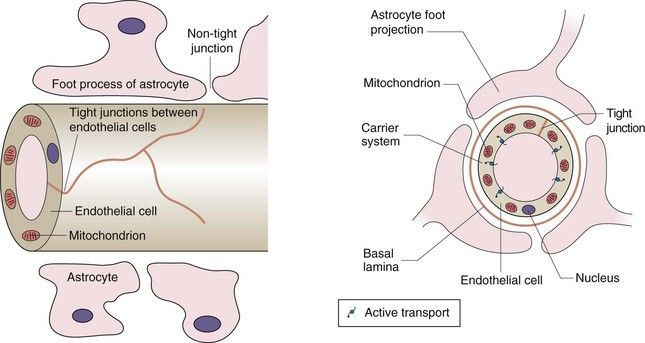
Fig. 2.7 The blood–brain barrier.
The barrier arises from the low number of membrane pores, the tight junctions between adjacent cells and the presence of efflux transporters that remove any drug that enters the endothelial cell. The presence of astrocytes is the stimulus for these changes in endothelial structure and function. Astrocytes are one of the several types of cells found in the CNS that make up the glia; they have numerous sheet-like processes and may provide nutrients to neurons.
 tight junctions between adjacent endothelial cells (capillaries are composed of an endothelial layer a single cell thick, with no smooth muscle),
tight junctions between adjacent endothelial cells (capillaries are composed of an endothelial layer a single cell thick, with no smooth muscle),

In addition, efflux transporters in the endothelial cells are an important part of the blood–brain barrier and return drug molecules back into the circulation, thereby preventing their entry into the brain and reducing effects in the central nervous system.
Water-soluble endogenous compounds needed for normal brain functioning, such as carbohydrates and amino acids, enter the brain via specific uptake transporters of the SLC superfamily (Table 2.1). Some drugs, for example levodopa, may enter the brain using these transport processes, and in such cases the rate of transport of the drug will be influenced by the concentrations of competitive endogenous substrates.
There is limited drug-metabolising ability in the brain and drugs leave by diffusion back into plasma, by active transport processes in the choroid plexus or by elimination in the cerebrospinal fluid. Organic acid transporters of the SLC superfamily (Table 2.1) are important in removing polar neurotransmitter metabolites from the brain.
Fetus
Lipid-soluble drugs can readily cross the placenta and enter the fetus. The placental blood flow is low compared to that in the liver, lung and spleen (Table 2.2); consequently, the fetal drug concentrations equilibrate slowly with the maternal circulation. Highly polar and very large molecules (such as heparin; see Ch. 11) do not readily cross the placenta. The fetal liver has only low levels of drug-metabolising enzymes so it is mainly the maternal elimination that clears the fetal circulation of drugs.
After delivery, the neonate may show effects from drugs given to the mother close to delivery (such as pethidine for pain control; see Ch. 19): such effects may be prolonged because the neonate now has to rely on his/her own immature elimination processes (Ch. 56).
Elimination
Elimination is the removal of drug from the body and may involve metabolism, in which the drug molecule is transformed into a different molecule, and/or excretion, in which the drug molecule is expelled in the body’s liquid, solid or gaseous ‘waste’.
Metabolism
A degree of lipid solubility is a useful property of most drugs, since it allows the compound to cross lipid barriers and hence to be given via the oral route. Metabolism is necessary for the elimination of lipid-soluble drugs from the body, because it converts a lipid-soluble molecule (which would otherwise be reabsorbed from urine in the kidney tubule; see below) into a water-soluble species capable of rapid elimination in the urine (often via an anion transporter).
Metabolism of the drug produces a new chemical entity, which may show different pharmacological properties from the parent compound:
 decrease in biological activity: the most common result of drug metabolism; arises from increased polarity which reduces receptor binding,
decrease in biological activity: the most common result of drug metabolism; arises from increased polarity which reduces receptor binding, increase in activity: the metabolite is more potent than the parent drug; a prodrug is an inactive compound that is converted by metabolism into the active molecular species,
increase in activity: the metabolite is more potent than the parent drug; a prodrug is an inactive compound that is converted by metabolism into the active molecular species,
The various steps of drug metabolism can be divided into two phases (Fig. 2.8). Although many compounds undergo both phases of metabolism, it is possible for a drug to undergo only a phase 1 or a phase 2 reaction, or for a proportion of the drug to be excreted unchanged. Phase 1 metabolism (oxidation, reduction and hydrolysis) is often described as preconjugation, because it produces a molecule that is a suitable substrate for a phase 2 or conjugation reaction. The enzymes involved in these reactions have low substrate specificities and can metabolise a wide range of drug substrates and other xenobiotics.
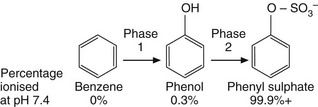
Fig. 2.8 The two phases of drug metabolism.
Phase 1 and phase 2 reactions are also called preconjugation and conjugation, respectively.
Phase 1
Cytochrome P450 is a superfamily of membrane-bound haemoprotein enzymes (Table 2.4). They are present in the smooth endoplasmic reticulum of cells (Fig. 2.9), particularly in the liver which is the major site of drug oxidation; the amounts in other tissues are low in comparison. The cytochrome P450 families CYP1–4 are involved in drug metabolism; the specific isoenzymes CYP2C9, CYP2D6 and CYP3A4 are involved in the phase 1 metabolism of approximately 10, 24 and 55% of drugs respectively.
Table 2.4
| Isoenzyme | Comments |
| CYP1A | Important for methylxanthines and paracetamol; induced by smoking |
| CYP2A | Limited number of substrates; significant inter-individual variability |
| CYP2B | Limited number of substrates |
| CYP2C | CYP2C9 is an important isoform; CYP2C19 shows genetic polymorphism |
| CYP2D | Metabolises numerous drugs; CYP2D6 shows genetic polymorphism |
| CYP2E | Metabolises alcohol |
| CYP3A | Main isoform in liver and intestine; metabolises 50–60% of current drugs |
| CYP4 | Metabolises fatty acids |
Human liver contains at least 20 isoenzymes of cytochrome P450. Families CYP1–4 are involved in drug metabolism.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


