- Programmed cell death is a normal physiological process in multicellular organisms and contributes to the efficient clearance of cells that have become damaged, aged, or simply surplus to requirements. It is a prominent feature of normal development but also contributes to tissue mass homeostasis throughout the life of the animal.
- Disrupting the normally harmonious equilibrium between cell proliferation and cell death can have a profound effect on the host. Excessive cell death can result in degenerative diseases, whereas cell death is invariably suppressed during the development of tumors.
- Apoptosis is a specialized form of programmed cell death that serves as a natural barrier to cancer development. Cancer cells have found ways to suppress apoptosis and thus avoid being killed.
- The machinery of apoptosis has been extensively described and comprises both an “extrinsic” and “intrinsic” pathway involving distinct upstream regulators and downstream effectors: the extrinsic pathway involves activation of death receptors (e.g. FAS), while the intrinsic pathway involves the release of cytochrome c from the mitochondria.
- In response to any of these apoptotic signals the final stages that lead to dismantling of the cell are executed by a subfamily of proteases known as caspases. Caspases are cysteine proteases that cleave hundreds of cellular proteins and ultimately lead to a series of morphological changes characteristic of apoptotic cell death.
- Cells in the final throws of apoptosis display “eat me” signals, like phosphatidylserine, that are recognized by phagocytes such as macrophages that then dispose of the corpse, without provoking an inflammatory response.
- A large and increasing number of proteins and protein families regulate apoptosis, including various tumors suppressor pathways and DNA damage responses on the proapoptotic side and growth factor signals on the antiapoptotic side.
- Inhibitors of apoptosis proteins (IAPs) are a family of proteins that are able to inhibit apoptosis by directly binding and inhibiting specific caspases. One of the mechanisms by which tumor cells are believed to acquire resistance to apoptosis is by overexpression of IAPs.
- The BCL-2 family of proteins comprises both proapoptotic and antiapoptotic members, the balance of which determines whether or not a cell commits suicide – apoptosis. These proteins have emerged as fundamental regulators of mitochondrial outer membrane permeabilization (MOMP), which is necessary for cytochrome c release. Overexpression of antiapoptotic BCL-2 family proteins is common to many human tumors, and confers resistance to apoptosis induced by standard anticancer therapies.
- The tumor suppressor p53, can prevent cells from becoming malignant by inducing growth arrest or apoptosis. There are several proapoptotic transcriptional targets of p53, such as BAX, NOXA, and PUMA that promote cytochrome c release from the mitochondria.
- Oncogenic proteins, such as c-MYC, that possess mitogenic action also induce apoptosis (or growth arrest) unless a survival signal is also received. This mechanism may operate as a “failsafe” to prevent cancer formation if an oncogene becomes deregulated.
- Most recently, another form of programmed cell death, termed “autophagy” (eating of self), has been postulated as a barrier to cancer development. Paradoxically, however, autophagy can also enable cancer cells to survive a variety of otherwise lethal stresses; the cancer cell lives to fight another day, by temporarily degrading cell organelles in lysosomes and reutilizing certain components until the stress is removed.
- Anticancer therapy can induce cellular senescence, differentiation, and/or cell death by apoptosis or by nonapoptotic mechanisms such as necrosis, autophagy, and mitotic catastrophe. However, since defects in apoptosis cause resistance to such therapy, restoring or activating apoptosis in tumors is an active area of cancer research.
Introduction
While I thought that I was learning how to live, I have been learning how to die.
Leonardo Da Vinci
All cells live under the shadow of death and when the inevitable happens, at least under normal physiological conditions, this usually occurs by apoptosis. A staggering 50 billion or more cells die each day in the human adult. Just to balance the books each one of us replaces around 70 kg of cells every year (enough to make a full-sized adult human clone – all be it minus some neurons).
Apoptosis, a characteristic form of programmed cell death often termed “cell suicide,” is a fundamental process that is essential for development, maintenance of tissue homeostasis, and the elimination of damaged cells. However, too much or too little apoptosis may lead to diseases such as neurodegenerative disorders, diabetes, or cancer. It is now widely accepted that putative cancer cells must avoid apoptosis in order for tumors to arise (see Chapters 6 and 7) and this is regarded as one of the “hallmark” features of cancer. This knowledge has fueled global research efforts into elucidating signal transduction pathways that mediate apoptosis as well as the mechanisms that have enabled cancer cells to avoid it (for example, through the loss of p53 tumor suppressor or overexpression of BCL-2 protein). For obvious reasons, a key objective in cancer research is to develop candidate drug molecules that can rekindle the suicidal urges of cancer cells and restore their sensitivity to apoptosis.
An Historical Perspective
There is only one ultimate and effectual preventive for the maladies to which flesh is heir, and that is death.
Harvey Cushing
Although studies on cell death are thought to have been performed centuries ago by Aristotle, and later by Galen, who described the regression of larval and fetal structures during development (and probably first used the term “necrosis”) it was not until after the formulation of the “cell theory” by Jacob Schleiden and Theodore Schwann in 1838 that nineteenth-century pathologists, in particular those of the “German school,” started to take interest in the process of cell death as a physiological phenomenon. Initially, it was appreciated that cells can die (in fact this was implicit from the time it was first realized that cells existed and were alive), but such death was assumed to be a passive response with cells as the victims of circumstances, such as poisons, and trauma, including death of the organism, largely beyond their control. This view began to change in the latter half of the nineteenth century, although it would be over a century later that cell death was first recognized by modern biology as a normal feature of multicellular organisms, and moreover, that this might be a process involving the active participation of the cell. In 1842, Carl Vogt suggested that cell death could be an important part of normal development based on observations in amphibian metamorphosis. This was followed by Rudolf Virchow, widely regarded as the father of modern pathology, who in 1858 described what he called “degeneration, necrosis and mortification.”
It was anatomist Walther Flemming who in 1885 proposed that cells might actually die spontaneously. During studies of regressing ovarian follicles, he observed nuclei that appeared to be breaking apart – a process he called “chromatolysis.” Intriguingly, Flemming’s sketches, appearing nearly a century before the concept of apoptosis was introduced and named, are probably the earliest clear example of cells undergoing apoptosis. In 1914, Ludvig Gräper suggested that mitosis would need to be balanced by processes such as “Flemming’s chromatolysis” that could keep proliferation in check. Every Yin needs its Yang. By the 1950s, embryologists such as Glucksmann had clearly described physiological cell death and Christian de Duve first proposed the concept of “cell suicide.” The term “programmed cell death” was introduced in 1964, to reflect the view that cell death during development was not an accidental occurrence but rather was part of a locally and temporally orchestrated plan.
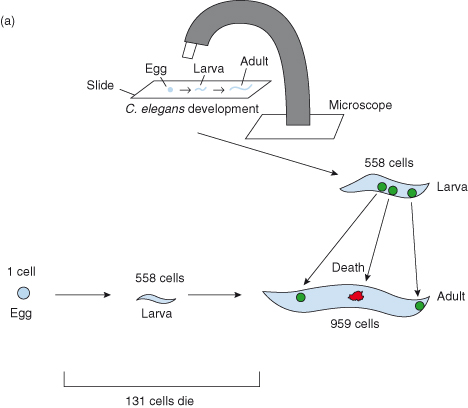
Probably the key event in recent times was when in 1971, John F. Kerr, Andrew Wyllie, and Sir Alistair Currie introduced the term “apoptosis” (from the Greek for falling of leaves) to describe this phenomenon of cell suicide, and thereafter founded the field of modern cell death research – although it was not until almost 20 years later that the idea that cells carried within them an intrinsic “suicide” program became generally accepted (see Box 8.1). A molecular explanation of physiological cell suicide was provided in the 1990s by H. Robert Horvitz (Nobel Prize winner – 2002) and colleagues who identified an intrinsic signaling pathway controlling the cell death of a group of specific neuronal cells during development in the worm Caenorhabditis elegans (Fig. 8.1).
Figure 8.1 Key experiment(s): Studies from H. Robert Horvitz, Nobel Prize winner (2002) and colleagues. The rediscovery of apoptosis as a generally important mechanism using Caenorhabditis elegans, the tiny, transparent nematode worm.
Caenorhabditis elegans is a 1-mm-long nematode (a roundworm) that lives in temperate soils. Research into the molecular and developmental biology of C. elegans was first pioneered by Sydney Brenner in Cambridge, UK in the 1960s. C. elegans is widely used as a model organism and has proved enormously useful in studying cellular differentiation and cell death. It was also the first metazooan to have its genome sequenced and surprised many researchers as it was found to contain more than 19 000 genes (more than 50% of the number of genes subsequently found in the human genome).
The relative simplicity of the organism has facilitated developmental studies and in the 1970s and 1980s John Sulston and Robert Horvitz, working with Brenner, traced the entire C. elegans embryonic cell lineage. We now know where every cell comes from and where they end up. In fact, the developmental fate of all 959 somatic cells has been mapped out. One aspect of the cell lineage particularly caught the attention of the Cambridge group: in addition to the 959 cells generated during worm development and found in the adult, another 131 cells are generated but are not present in the adult. These cells are absent because they undergo programmed cell death (see Nobel lecture by Horvitz).
a Cell lineage studies in the developing worm (John Sulston and Robert Horvitz). Newly hatched C. elegans larvae were placed on a glass microscope slide dabbed with a sample of the bacterium Escherichia coli (nematode food). Then, using Nomarski differential interference contrast optics, individual cells within the living animal could be closely observed and followed as they migrated, divided, and died. In this way, the fate of every single cell from the larval stage to the adult worm could be determined and a cell lineage map was generated.
Subsequently, the pattern of cell divisions between the single-celled fertilized egg and the newly hatched larva were tracked. This was far more difficult than tracking cell fates in larvae, in part because the process of embryonic morphogenesis involves a major cellular rearrangement of a ball of cells to generate a worm-shaped larva. Imagine watching a bowlful of hundreds of grapes, trying to keep your eye on each grape as it and many others move! Nevertheless, all 558 nuclei were followed.
Together, these studies defined the first, and to date only, completely known cell lineage of an animal.
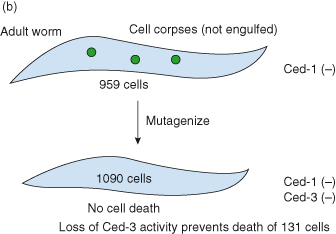
b Identifying cell death genes. The next step was to identify the genes responsible for causing or preventing programmed cell death during worm development. Researchers mutagenized a ced1 (cell death abnormal) mutant worm in which the process of engulfment, or phagocytosis, that normally removes dying cells from the body of the animal, is defective. In ced1 mutants, although programmed cell death still occurs, dead or dying cells are not engulfed and so cell corpses persist and can be easily visualized in living individuals using Nomarski optics. By mutagenizing ced1 animals, it was hoped that mutants possessing abnormalities in the pattern of programmed cell deaths (as seen with Nomarski optics) would be generated. In this way, mutants could be identified in which the process of programmed cell death had not been initiated or in which the pattern of programmed cell deaths was altered.
Indeed, a mutant was found in which no cell corpses could be seen. The gene defined by this mutant was called ced3. It was shown that if ced3 activity is reduced or eliminated by mutation, essentially all 131 cells that normally die instead survive–hence the absence of cell corpses. Ced-3 protein, it was later discovered, was in fact a caspase.
A second mutant was then discovered (using different mutant animals) that prevented cell death. These worms proved to be defective in a new gene with properties essentially identical to those of ced3. This killer gene was named ced4. Some years later, in 1997, a protein similar to Ced-4 was identified, called Apaf-1 (apoptotic protease-activating factor), with a domain with significant similarity to Ced-4. APAF-1 is a proapoptotic human protein similar in both sequence and function to the C. elegans programmed cell death killer protein Ced-4.
In a similar manner, the ced9 gene was discovered, and was later shown to be homologous to the human proto-oncogene, BCL2, whose protein product serves to protect cells against cell death.
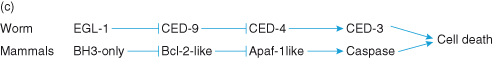
c Genetic pathway for programmed cell death. From these studies, the core molecular genetic pathway for programmed cell death in C. elegans has been identified which shows great similarity to the mammalian pathway: Egl-1 (egg-laying abnormal) is similar to mammalian “BH3-only” proapoptotic proteins; Ced-9 is similar to mammalian antiapoptotic Bcl-2 protein; Ced-4 is similar to Apaf-1 (important for the activation of procaspase-9 on the apoptosome); Ced-3 is similar to caspases.
Apoptosis in Context
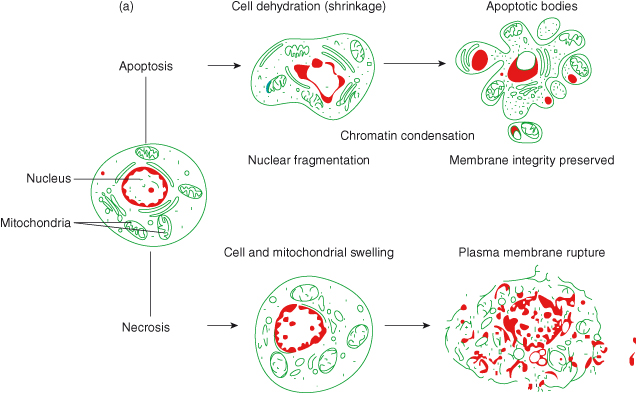
Apoptosis is an orchestrated cell death process in which the entire cell is dismantled within the orderly context of membrane-enclosed vesicles, thereby preventing any untoward spillage of intracellular components from the dying cell that might otherwise provoke an immune response. In fact, the risk of inadvertent leakage of any noxious contents is avoided by the rapid clearance of the apoptotic corpses and their membrane-bound fragments through phagocytosis by macrophages and neighboring cells (Fig. 8.2). In contrast, cell death by necrosis incites an inflammatory response, because the cell and its organelles swell and rupture, showering cellular contents into the surrounding tissue. Necrosis is often seen as a more passive cell response to profound physical, chemical, or genotoxic insults.
Figure 8.2 Cell death by apoptosis versus necrosis.
a Schematic representation of cell death by apoptosis and necrosis. Once the apoptotic program has been initiated within a cell, several morphological characteristics can be seen using high-power microscopy: the cell loses contact with its neighbours; chromatin condenses at the edge of the nucleus; DNA fragmentation, cell shrinkage, and dilatation of the endoplasmic reticulum occur. “Blebs” appear on the cell surface, leading to budding of cell membrane and packaging of cellular components into vesicles – known as “apoptotic bodies.” These apoptotic bodies are phagocytosed by macrophages and neighboring cells, thus avoiding an inflammatory response. In contrast, death by necrosis proceeds with loss of function of mitochondria and endoplasmic reticulum resulting in a dramatic breakdown of energy supply. The outcome is cellular, nuclear, and organellar swelling. The ultimate rupture of the plasma membrane allows the release of lysosomal enzymes, which attack neighboring cells and surrounding tissue, thus triggering an inflammatory response. Based on image from Google Images.
b Scanning electron micrograph of liver cells dying by apoptosis. Note the obvious “blebs” on the cell surface. Image reproduced with permission from Science Photo Library.
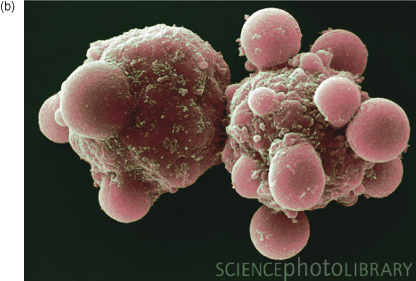
The elimination of cells by apoptosis is an essential feature of normal physiology and is observed throughout animal development and tissue homeostasis in the adult. During development, apoptosis occurs during the sculpting of somatic structures – for example, the loss of interdigital webs during the formation of the digits, and the hollowing out of solid structures to create lumina such as the gut. In the developing nervous system, only half of the neurons formed receive sufficient survival signals from their target cells; the remaining neurons die by apoptosis. Although this appears an inefficient and wasteful process, in fact this developmental mechanism is believed to be vital for ensuring the correct innervation of target cells by appropriate neurons. Similarly, during development of the immune system, up to 95% of all cells die by apoptosis because they make unproductive or autoreactive antigen receptors. Apoptosis in development is often referred to as “programmed cell death.” An example of when the regulation of apoptosis plays a key role in the adult is following childbirth – in preparation for lactation there is growth of breast tissue (proliferation > apoptosis). However, following lactation, dramatic regression of tissue occurs by apoptosis.
Apoptosis continues throughout the life of an animal in order to maintain tissue homeostasis, that is, a balance between cell proliferation and cell death. In fact, in an adult human, billions of cells die every hour! Maintaining tissue homeostasis is particularly important in those tissues that have a high cell turnover, such as epithelial (e.g. skin epidermis and gut) and hematopoietic tissues. It is therefore not surprising that perturbing the balance between cell proliferation and cell death can have a profound effect on the host. For example, excessive apoptosis is associated with degenerative diseases such as Alzheimer disease, spinal muscular atrophy, Huntington disease, and Parkinson disease. Conversely, suppression of apoptosis is essential during the development of tumors – although highlighted at various points throughout the chapter. This deleterious effect is discussed in Chapter 6 in relation to oncogenes, the so-called “cancer-causing” genes.
Here we will describe the process of apoptosis, the signal transduction pathways controlling this form of cell death, and highlight ways in which normal apoptosis regulation becomes derailed in cancer. We will also discuss other forms of programmed cell death, including the more recently described “autophagy” that appears to play a role both as a barrier to cancer and paradoxically as a means by which cancer cells could survive stress. We will conclude by discussing the potential of targeting cell death pathways as cancer therapies.
Apoptosis as a Barrier to Cancer Formation
As is the generation of leaves, so is that of humanity.
The wind scatters the leaves on the ground, but the live timber
Burgeons with leaves again in the season of spring returning.
So one generation of men will grow while another dies.
Homer
As described in earlier chapters, cancers arise as a result of accumulation of multiple genetic and epigenetic lesions that allow cells to proliferate uncontrollably irrespective of exogenous mitogens, resist apoptosis, recruit a blood vasculature, invade surrounding tissues, and eventually metastasize – vide infra the hallmark features of cancer.
In normal adult tissues, apoptosis ensures orderly homeostasis by eliminating cells that are damaged (e.g. DNA damage, hypoxia, nutrient limitation) or displaced (cells that have moved out of their normal environment – ”anoikis,” see Chapter 12). However, the acceptance of apoptosis as one of the central tumor suppressive mechanisms has gradually effloresced over the last decade. It is now unquestioned that evolving cancer cells must avoid cell death for tumors to arise. We now know that cells acquiring a growth-promoting mutation, exemplified by overexpression of the oncogenes c-MYC, E1A, or E2F, become “sensitized” to apoptosis (see Chapter 6). The importance of this cannot be overestimated, as such cells can be eliminated from the body and thus avoid becoming cancerous cells. The notion that a growth-deregulating oncogenic mutation in vivo could also possess an “in-built” tumor suppressor function to hinder expansion of potentially malignant cells is one of the most important concepts in cancer biology (and is featured in several chapters; see Chapters 6, 7, and 10). In particular, through the development of regulatable mouse models (see “Mouse models of tumor progression” in Chapter 6), the importance of apoptosis serving as a barrier to cancer formation is all too clear – when apoptosis is blocked, tumor progression is accelerated. As the best-studied example of an oncogene with intrinsic tumor suppressor activity, the various signaling pathways that c-MYC may activate to promote apoptosis are described later.
Proto-oncogenes that specifically regulate apoptosis and which are often deregulated in human cancers include members of the BCL2 family, the BCL2 oncogene being the first identified by the study of chromosomal translocations in human lymphoma (see Chapter 3, Box 3.3). The BCL-2 family is now known to include a large number of proteins with either anti- or proapoptotic activities which are described later. If expression of antiapoptotic BCL-2 family members is increased, or that of proapoptotic members lost, then cells are able to survive and consequently become vulnerable to other cancer-causing mutations, in particular those which increase proliferation (e.g. MYC). Instead of dying, as they would normally do, these cells are now able to survive and give rise to cancer.
It is clear that highly conserved pathways regulating cellular growth/replication and apoptosis are coupled at numerous levels, even under normal physiological conditions, and that this plays a key role in the regulation of appropriate cell numbers and organ size during development and in maintaining these in the adult. In addition to the role of c-MYC, E2F, and others in this process, studies in the fruitfly (Drosophila) have revealed another important signaling pathway involving the large tumor suppressor LATS/WARTS protein kinase, which alongside two other proteins, Hippo and Salvador, is also a regulator of both cell-cycle exit and apoptosis (see Chapter 5; Figs 5.23 and 5.24). At least two mammalian homologs of LATS/WARTS are known, LATS1 and LATS2, and disruption of LATS1 or in proteins, such as the Hippo homolog MST2 or the MATS1 (Mob as tumor suppressor), required to activate LATS1 prevent apoptosis and increase tumor formation in mice. ABL can also exert dual roles, but in this case may be determined by cellular location. In the cytoplasm, ABL induces replication (the BCR–ABL protein in chromic myelogenous leukemia is confined to the cytoplasm), whereas in the nucleus ABL promotes apoptosis (see Chapter 6). Moreover, some of these have now been shown to be disrupted in human cancers. It is worth noting here that an analogous role may be played by senescence (see Chapters 6 and 9) as a barrier to RAS-induced tumorigenesis.
As this chapter focuses mainly on the molecular pathways specifically involved in apoptosis, we urge the reader to look at Chapters 4–7 for a broader overview of the regulation of cell cycle, growth, and the contributions of oncogenes and tumor suppressors, cell death (or its avoidance) and cancer.
Lastly, apart from elucidating mechanisms that have enabled cancer cells to escape apoptosis, it also provides an avenue for future development of candidate drug molecules to target cancer (see last section on “Exploiting cell death (and senescence) in cancer control”).
Apoptosis Versus Necrosis
It is possible to state as a general principle that the mesodermic phagocytes, which originally (as in the sponges of our days) acted as digestive cells, retained their role to absorb the dead or weakened parts of the organism as much as different foreign intruders.
Metchnikoff
Apoptosis typically affects single cells that are aged, dysfunctional, or damaged by extermal stimuli. Unlike necrosis, it is an active, energy-requiring process leading to a characteristic series of morphological changes that accompany the degradation of the cell. Key differences between the two forms of cell death are described below.
During apoptosis, gross morphological changes – as shown schematically in Fig. 8.2(a) – can been seen under a microscope. Early features of apoptosis occur within minutes of the apoptotic trigger, and during this phase mitochondria, lysosomes, and cellular membranes remain fully intact. These features include: chromatin condensation, DNA fragmentation (multiples of 180 bp), cell shrinkage, and dilatation of the endoplasmic reticulum.
Later features of apoptosis are completed within hours, depending on cell type and tissue: budding of cell membrane (“blebs”) leading to the packaging of cellular components into vesicles – known as “apoptotic bodies,” which are phagocytosed by macrophages and neighboring cells, thus avoiding an inflammatory response. Figure 8.2(b) shows a scanning electron micrograph of apoptotic cells with obvious “blebs” on the cell surface.
In contrast, necrosis affects groups of cells or whole tissue after extended damage induced by external stimuli such as trauma, ischemia, and high-dose irradiation. This energy-independent form of cell death occurs over 12–24 hours, with loss of function of mitochondria and endoplasmic reticulum, resulting in a dramatic breakdown of energy supply leading to cellular, nuclear, and organellar swelling, and ultimately rupture of the plasma membrane (Fig. 8.2a). This final event allows the release of lysosomal enzymes which attack neighboring cells and surrounding tissue, thus triggering an inflammatory response. In contrast to apoptosis, there is nonspecific degradation of DNA.
Cell Death by Necrosis – Not Just Inflammatory
Although necrosis has been acknowledged for many years as a form of cell death in which cells become bloated and explode, until recently it has not featured as being of particular relevance to tumorigenesis.
As mentioned above, necrosis completely differs from apoptosis, in which the dying cell contracts into a tiny corpse that is soon “eaten” by neighboring cells. Once exploded, necrotic cells release their contents into the local tissue microenvironment and induce a proinflammatory response, including recruiting of inflammatory cells such as neutrophils and macrophages. These inflammatory cells serve an important function in damaged tissue by dealing with potential pathogens, and then ultimately phagocytes (e.g. macrophages) remove the necrotic debris, terminating the inflammation and allowing tissue repair. However, in the context of tumor growth, there is increasing evidence that immune inflammatory cells can be actively tumor promoting (touched upon in the section on “The IAP family” and in Chapter 13) being capable of inducing angiogenesis, cancer cell proliferation and invasiveness.
In addition, necrotic cells can release regulatory factors like interleukin 1α (IL-1α), which can directly stimulate neighboring cells to proliferate. In this scenario, tumors in which necrotic cell death is supposedly beneficial in counterbalancing cancer-associated hyperproliferation, may ultimately do more damage by recruiting inflammatory cells that promote growth of the surviving tumor cells. Thus, tumors can appear as wounds that do not heal – chronic cell death leads to inflammation that does not resolve, and instead enhances tumor growth.
The Pathways to Apoptosis
In general, there are two pathways that trigger apoptosis – the extrinsic and intrinsic (mitochondrial) apoptotic pathways. The extrinsic pathway, otherwise known as the “death receptor pathway” is activated by the engagement of death receptors on the cell surface. In contrast, the intrinsic pathway involves the release of cytochrome c (and other proteins) from the mitochondria. Whichever pathway is taken (Fig. 8.3), both lead to the activation of various caspases – enzymes responsible for the demise of the cell. These crucial enzymes are discussed later in their own section. It is important to emphasize that in vertebrates the majority of apoptosis proceeds through the mitochondrial pathway.
Figure 8.3 The two pathways of apoptosis: death receptor (extrinsic) and mitochondrial (intrinsic) pathways. Here, the death receptor pathway is triggered following binding of FAS ligand (FASL) to the FAS receptor (FAS). FASL binding induces clustering of FAS, which in turn recruits the adaptor protein FADD (FAS-associated death domain) to form a complex called the death-inducing signaling complex (DISC). DISC then recruits and activates the initiator procaspase-8. Subsequently, activated procaspase-8 triggers a caspase cascade, activating downstream executioner caspases, such as caspase-3 and caspase-7, that ultimately kill the cell. However, caspase-8 can also activate the proapoptotic protein BID (by cleavage to become truncated tBID), which promotes release of cytochrome c from the mitochondria – the “intrinsic” pathway. The mitochondrial pathway can be triggered by a variety of cellular stresses (e.g. DNA damage, hypoxia, depleted survival factors or deregulated oncogenes). Once released into the cytosol, cytochrome c associates with APAF-1 to create the apoptosome, a complex that activates procaspase-9. In the presence of cytochrome c and the nucleotide dATP/ATP, procaspase-9 is autocatalytically activated and can now go on to activate downstream executioner caspases, such as caspase-3 and caspase-7. Other proteins that are released from the mitochondria include Smac/DIABLO (and probably Htra2/Omi), which bind to and inhibit inhibitors of apoptosis proteins, thus preventing caspase-9 and caspase-3 inhibition.
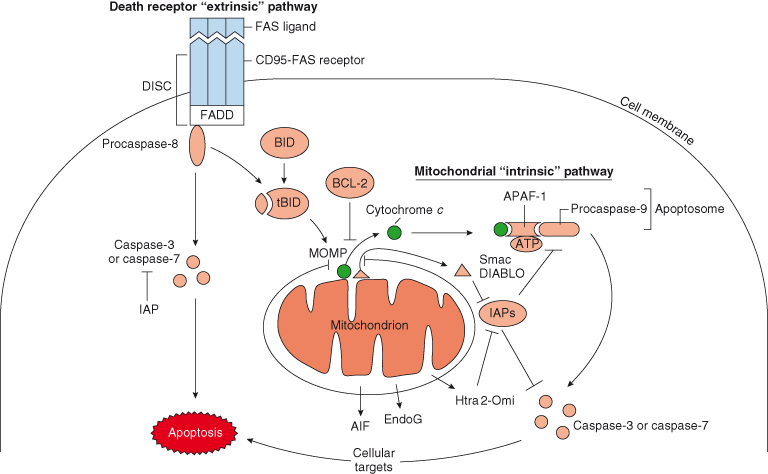
Figure 8.4 Functional domains of the APAF-1 protein. APAF-1 contains an N-terminal caspase recruitment domain (CARD) by which it interacts with procaspase-9, a nucleotide binding domain (NBD), and a long C-terminal extension containing 13 repeats of the WD40 motif. The WD40 motif is a conserved protein domain approximately 40 residues long that has a characteristic tryptophan–aspartate motif, and is thought to negatively regulate APAF-1.

Death Receptor Pathway (Extrinsic)
This pathway is initiated by the binding of ligands to cell surface receptors and as such are the antithesis to growth factors and their receptors. However, the actual role played by this mechanism in normal tissue mass homeostasis is unclear, and most evidence suggests that this system is primarily involved in immune cell killing of viral infected host cells and in other pathological states. Extracellular hormones or agonists that belong to the tumor necrosis factor (TNF) superfamily, including TNF-α, FAS/CD95 ligand, and APO2 ligand/TRAIL, bind to their specific corresponding receptors (TNF/NGF receptor family), such as TNFR1, FAS/CD95, and APO2, respectively. The resultant ligand–receptor binding activates downstream signaling cascades. For example, as shown in Fig. 8.3, binding of FAS ligand (FASL) to the FAS receptor (FAS) induces clustering of FAS, which in turn recruits the adaptor protein FADD (FAS-associated death domain) to form a complex called the death-inducing signaling complex (DISC). DISC then recruits and activates the initiator procaspase-8, probably by bringing the procaspases close enough in proximity (“close proximity” model) so that they can cleave each other. Subsequently, these activated initiator caspases trigger a caspase cascade, activating downstream executioner caspases, such as caspase-3 and caspase-7, that ultimately kill the cell.
When large amounts of caspase-8 are formed at the DISC, then apoptosis proceeds via direct cleavage of procaspase-3 independently of the mitochondria (see mitochondrial pathway). However, caspase-8 can also activate the proapoptotic BCL-2 family protein BID, which promotes release of cytochrome c from the mitochondria – the “intrinsic” pathway described below (Fig. 8.3). This “crosstalk” between the death receptor and mitochondrial pathways can occur in cells when DISC formation and active caspase-8 are insufficient to activate procaspase-3 independently of the mitochondria. Cells in which the extrinsic pathway induces apoptosis only when MOMP is intact are referred to as type II cells, whereas cells in which this is not required and the extrinsic pathway alone is sufficient to induce cell death are referred to as type I.
Apoptotic cell death mediated via death receptors is critical for normal immune system function, for example, mutations in FAS and FASL ligand in humans can lead to a complicated immune disorder known as the autoimmune lymphoproliferative syndrome (ALPS), a similar phenotype to that seen in mice with Fas and FasL mutations.
Importantly, in some cell types, apoptotic response to anticancer therapy implicates the death receptor FAS (see last section). Various anticancer drugs can activate the death receptor pathway by enhancing the expression of FAS and FASL. Interaction of FASL with FAS at the cell surface defines an autocrine/paracrine pathway similar to that observed in activation-induced cell death in T lymphocytes. However, it is possible that FASL is not crucial for drug-induced apoptosis since apoptosis is not suppressed by antagonist antibodies or molecules that prevent FASL binding to FAS. It is likely that certain anticancer drugs exert their apoptotic effects by inducing clustering of FAS receptor at the cell surface of tumor cells in the absence of FASL.
The Mitochondrial Pathway (Intrinsic)
It is now known that the mitochondria play a central role in most apoptotic cell death in vertebrate cells. The key and defining event in the mitochondrial pathway is MOMP (mitochondrial outer membrane permeabilization), as a result of which, various proteins normally confined to the mitochondrion are released and trigger the subsequent dismantling of the cell (Fig. 8.3). Cytochrome c was the first of these mitochondrial proteins to be detected in the cytoplasm of apoptotic cells. At first, this observation was rather puzzling, not least because cytochrome c is an essential protein in energy production within cells and is usually located inside the mitochondria (in the space between the inner and outer mitochondrial membranes). Subsequently, however, the role of cytochrome c in activating apoptosis was confirmed by two findings: the first was the identification of its downstream binding partner, APAF-1 (apoptotic protease-activating factor 1) present in the cytosol (described next under “The apoptosome”), and the second was the demonstration that the antiapoptotic protein BCL-2 (described later) inhibits cell death by preventing release of cytochrome c from the mitochondria.
Pressaging the success of Downton Abbey on both sides of the Atlantic, Doug Green, in a review article in the journal Science, posited a very apt analogy to describe the mitochodrial pathway – ”we have an upstairs/downstairs situation where at first pass most of the aristocratic decisions are made before MOMP (upstairs) and the workmanlike consequences occur thereafter (downstairs).” Implicit in this is the notion that executive decisions and balancing of factors for and against apoptosis are made before MOMP. Once MOMP has been “signed off,” the order for apoptosis is largely carried out regardless. This does not mean that there is no debate about matters after MOMP – the various interactions between other mitochondrial proteins and IAPs (see below) can modulate the decision to commit apoptosis. A key question is: what happens to promote MOMP and thence the release of cytochrome c and other proteins from mitochondria?
Without a doubt, the BCL-2 family of proteins are key regulators of MOMP and will be discussed in more detail later under their own heading, but it is worth continuing the upstairs/downstairs analogy at this point. As we will see, BAX and BAK are the “butlers” of this piece; they operate above and below stairs and most importantly we know by literary convention that they are culpable. We also know something of the motive; the normally affable BAX and BAK are driven to “commit MOMP” by imbalances between pro- and antiapoptotic BCL-2 family members upstairs. Stress of various kinds can trigger activity of proapoptotic (BH3-only) proteins in part by removing the calming embrace of antiapoptotic BCL-2 and BCL-XL. The nature of the upstairs activating signals that lead to changes in expression and/or activation of various BCL-2 family members will vary depending on cell type and what stresses the cell is under, for example, hypoxia, depleted survival factors, DNA damage, or if the cell has acquired an oncogenic mutation (e.g. in c-MYC leading to deregulated expression).
Once MOMP occurs, cytochrome c can associate with APAF-1 in the cytoplasm to create the apoptosome, a complex that activates procaspase-9. Since formation of the apoptosome plays a crucial role in mediating apoptosis, it deserves a more thorough explanation (see below). In the presence of cytochrome c and the nucleotide dATP/ATP, procaspase-9 forms oligomers and becomes autocatalytically activated; active caspase-9 can now go on to activate downstream executioner caspases, such as caspase-3 and caspase-7 (Fig. 8.3), by cleavage at specific sites.
The regulation of apoptosis downstairs is complex and involves numerous activation steps and also protein stability. Several different inhibitors of caspases, the inhibitor of apoptosis proteins (IAPs), have been described that inhibit apoptosis by targeting caspases for proteasome degradation. Conversely, various proteins released from the mitochondrial intermembrane space, alongside cytochrome c (Fig. 8.3), allow MOMP to circumvent the action of the apoptosis-inhibiting IAPs. Thus, MOMP releases two inhibitors of IAPs, DIABLO/Smac (second mitochondria-derived activator of caspase) and Omi/HtrA2. In fact, other important proteins are released by MOMP, including two which can contribute to apoptosis even in the absence of caspase activation – AIF (apoptosis-induction factor), involved in chromatin condensation and large-scale DNA degradation, and endonuclease G (EndoG), which might aid the caspase-activated DNase (CAD) in nucleosomal DNA fragmentation. In some circumstances MOMP may even result in release of a small proportion of procaspase molecules.
The Apoptosome – ”Wheel of Death”
The three-dimensional structure of the apoptosome has now been solved using cryoelectron microscopy technology. The structure has given insight into how the apoptosome assembles, how it might activate procaspase-9 and why activation of this procaspase is distinct from the conventional caspase activation mechanism (that is, proteolytic cleavage of effector caspase), which is discussed in more detail in the below.
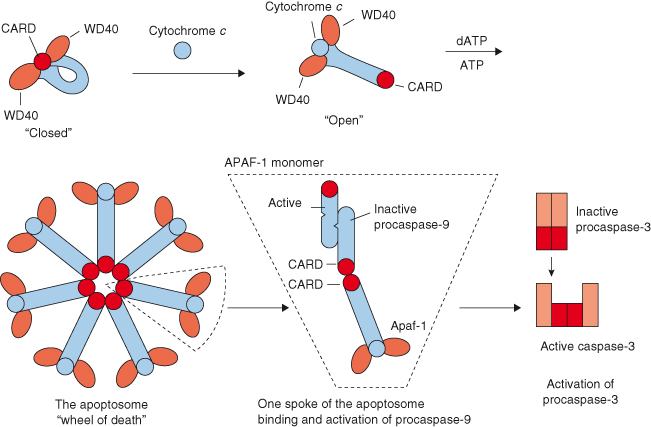
Once cytochrome c is released from mitochondria into the cytosol, it associates with APAF-1 to create the apoptosome, a complex that activates procaspase-9. In mammalian cells, APAF-1 is cytosolic and, importantly, its activity is restrained by its long C-terminal extension containing 13 repeats of the WD40 motif (Fig. 8.4). The WD40 motif is a conserved protein domain approximately 40 residues long that has a characteristic tryptophan–aspartate motif. In an individual APAF-1 molecule, two groups of WD40 repeats in the C-terminal region are thought to keep the protein inactive until cytochrome c engages the repeats (Fig. 8.5). After association with cytochrome c, APAF-1 switches from a rigid conformation (“closed”) to a more flexible one (“open”) such that the nucleotide dATP/ATP binding activity is greatly facilitated. This binding triggers formation of the active seven-span symmetrical wheel-like structure (“wheel of death”) – the apoptosome – via interaction among the N-terminal caspase recruitment domains (CARD) of the individual APAF-1 molecules (Fig. 8.5). The apoptosome subsequently recruits procaspase-9 into its central hub through CARD–CARD domain interaction between procaspase-9 and APAF-1 molecules, which brings about a conformational change of procaspase-9. This enzyme is now in its active form and can go on to activate downstream executioner caspases, such as caspase-3 and caspase-7, which will eventually lead to cell death.
Figure 8.5 Formation of the apoptosome. In an individual APAF-1 molecule, two groups of WD40 repeats in the C-terminal region are thought to keep the protein inactive until cytochrome c engages the repeats. Association with cytochrome c causes APAF-1 to convert from a “closed” conformation to a more “open” one, thus allowing the nucleotide dATP/ATP binding activity to be greatly facilitated. This binding triggers formation of the active seven-span symmetrical “wheel of death” – the apoptosome – via interaction among the N-terminal caspase recruitment domains (CARD) of the individual APAF-1 molecules. The apoptosome subsequently recruits procaspase-9 into its central hub through CARD–CARD domain interaction between procaspase-9 and APAF-1 molecules. An inactive procaspase-9 monomer on one “spoke” of the apoptosome is thought to recruit another monomer to create a dimer with a single active site. This active caspase activates downstream executioner caspases, such as caspase-3 and caspase-7.
Caspases – the Initiators and Executioners of Apoptosis
Once apoptotic signals are received by the cell, the final stages that lead to dismantling of the cell are executed by a subfamily of proteases known as caspases. Caspases are highly selective cysteine proteases that have a preference for cleaving proteins after aspartate residues. This specificity ensures that apoptosis is primarily a set of limited proteolytic cleavages, and not a generalized degradative process. Caspase-dependent cleavage is of a defined subset of cellular proteins comprising around 1 in 20 of all proteins (1000 or so) that together culminate in the various morphological changes characteristic of apoptotic cell death.
It is important to note that caspases play roles in other processes, including those involving cellular remodeling such as spermatid individualization, macrophage and skeletal muscle differentiation, cornification of skin, and erythropoiesis. Understanding how this is confined and apoptosis avoided under these circumstances is an important area of research. In some cases this involves specific caspases restricted to certain tissues (such as caspase-14 in skin) that may also be activated by routes separate from apical caspases. Interactions with the ubiquitin–proteasome system (see Chapter 11) are also believed to be important regulators of caspases.
Caspases can be grouped into two categories: initiator caspases and effector (or executioner) caspases. As mentioned earlier (see Fig. 8.3), initiator caspases (e.g. caspases-2, -8, and -9) are activated as they bind to their appropriate adaptor molecules (APAF-1, FADD; see below), after which they cleave and thereby activate downstream (“effector”) procaspases and various proteins. The downstream “effector” (or executioner) caspases (e.g. caspases-3, -6, and -7) go on to degrade many cellular substrates (Box 8.2) that result in the characteristic features of apoptotic cell death.
Caspases are synthesized and stored as inactive precursors, procaspases, in order to protect the cell from untimely proteolytic events. Each procaspase consists of a prodomain at its N-terminus, a large subunit, and a small subunit (Fig. 8.6). The length of the prodomain varies among caspases (Table 8.1) – long prodomains are characteristic of initiator caspases and contain a death effector domain (DED in procaspases-8/10) or caspase recruitment domain (CARD in procaspase-9). These domains mediate the homophilic interactions (i.e. CARD–CARD and DED–DED) between procaspases and their adaptor proteins (e.g. APAF-1 with procaspase-9; FADD with procaspase-8). An example of this can be seen in Fig. 8.5 during formation of the apoptosome.
Table 8.1 Procaspases subgrouped on the basis of domain composition
| Long prodomain (DED, CARD) – apoptotic initiator type | Caspases-2,8,9,10 |
| Short prodomain – apoptotic effector (executioner) type | Caspases-3,6,7 |
| Long prodomain (CARD) – cytokine activator type | Caspases-1,4,5 |
The human genome encodes 11 caspases, and these can be divided into subgroups depending on inherent substrate specificity, domain composition, or presumed role in vivo.
Figure 8.6 Initiator and executioner caspases. Caspases are highly selective cysteine proteases that have a preference for cleaving proteins after aspartate residues (Asp). Initiator procaspases such as procaspase-8 and procaspase-9 possess long prodomains which contain a protein interaction domain, such as the death-effector domain (DED) and caspase-recruitment domain (CARD), respectively. These domains mediate the homophilic interactions (i.e. CARD–CARD and DED–DED) between procaspases and their adaptor proteins (e.g. APAF-1 with procaspase-9; FADD with procaspase-8) and are indispensible to the activation of initiator caspases. Executioner procaspases, such as procaspase-3, have short prodomains which seem to inhibit caspase activation. Procaspase-3 is activated by initiator caspases: it is cleaved at specific Asp residues to yield a short inhibitory prodomain, a large and a small subunit, termed homodimer p20 and p10, respectively. Subsequently, p20 and p10 homodimers interact in a heterodimer – the association of two heterodimers forms the proteolytic tetramer with the two adjacent p10 subunits surrounded by two large subunits.
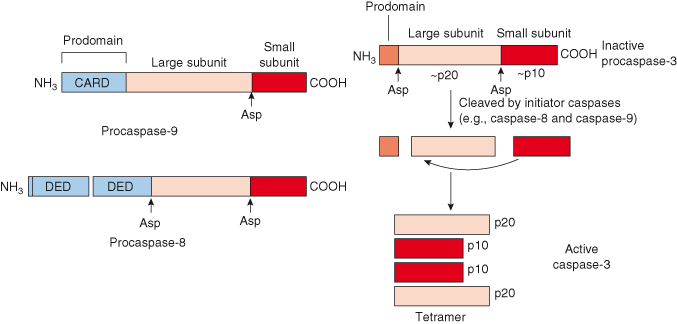
It is now recognized that activation of initiator caspases occurs following oligomerization of adaptor proteins (APAF-1, FADD) which in turn leads to recruitment and autoactivation of procaspases (procaspase-8 and -9, respectively). This mode of activation differs from that required for executioner procaspases, which typically require proteolytic cleavage. Executioner procaspases (procaspases-3, -6, and -7) have short prodomains which seem to inhibit caspase activation. These caspases exist constitutively as homodimers – both before and after activation cleavage. Executioner procaspases are activated by initiator caspases; for example, procaspase-3 and -7 can be activated by caspases-6, -8, -9, and -10. During activation, the executioner procaspase is cleaved at specific Asp residues to yield a short inhibitory prodomain, a large and a small subunit, termed homodimer p20 and p10, respectively (Fig. 8.6). Subsequently, p20 and p10 homodimers (refers to subunit size, in kDa) interact in a heterodimer; the association of two heterodimers forms the proteolytic tetramer with the two adjacent p10 subunits surrounded by two large subunits. The catalytic activity of an executioner caspase is increased by several orders after such cleavage.
Unlike executioner procaspases, our understanding of how initiator procaspases are activated is far from complete, but appears to require an appropriate scaffold in most cases. With regard to the activation of procaspase-9 by the apoptosome (described previously), two models have been proposed: the “induced proximity” model and the “proximity-induced dimerization” model. The first model states that the initiator procaspases autoprocess themselves when they are brought into close proximity. However, the mechanism underlying such caspase activation is not known. The second model states that procaspase-8 and procaspase-9 are activated on dimerization, which is facilitated by the apoptosome and DISC complexes, respectively. Procaspase-9 can be activated without processing, apparently because of the flexibility allowed by the unusually long linker between its large and small subunits. An inactive procaspase-9 monomer on one spoke of the apoptosome is presumed to recruit another monomer (homodimerization) in an antiparallel fashion to create the asymmetric dimer having a single active site (see Fig. 8.5). This is a unique feature as only one of the two constituent polypeptides becomes active.
Importantly, most of the active caspase-9 remains complexed to APAF-1 and the released caspase-9 has minimal activity, thus indicating that APAF-1 is not merely a scaffold but within the apoptosome acts as a holoenzyme. In fact, after associating with the apoptosome, there is a dramatic increase in the catalytic activity of caspase-9 (up to 2000-fold) of both processed (cleaved) and unprocessed caspase-9. Thus, for caspase-9 at least, activation has little to do with cleavage; rather, it occurs through apoptosome-mediated enhancement of the catalytic activity of caspase-9.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


