28
Catabolism of Proteins & of Amino Acid Nitrogen
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe protein turnover, indicate the mean rate of protein turnover in healthy individuals, and provide examples of human proteins that are degraded at rates greater than the mean rate.
Describe protein turnover, indicate the mean rate of protein turnover in healthy individuals, and provide examples of human proteins that are degraded at rates greater than the mean rate.
![]() Describe the events in protein turnover by both ATP-dependent and ATP-independent pathways, and the roles in protein degradation of ubiquitin, cell surface receptors, circulating asialoglycoproteins, and lysosomes.
Describe the events in protein turnover by both ATP-dependent and ATP-independent pathways, and the roles in protein degradation of ubiquitin, cell surface receptors, circulating asialoglycoproteins, and lysosomes.
![]() Indicate how the ultimate end products of nitrogen catabolism in mammals differ from those in birds and in fish.
Indicate how the ultimate end products of nitrogen catabolism in mammals differ from those in birds and in fish.
![]() Illustrate the central roles of transaminases (aminotransferases), of glutamate dehydrogenase, and of glutaminase in human nitrogen metabolism.
Illustrate the central roles of transaminases (aminotransferases), of glutamate dehydrogenase, and of glutaminase in human nitrogen metabolism.
![]() Use structural formulas to represent the reactions that convert NH3, CO2, and the amide nitrogen of aspartate into urea.
Use structural formulas to represent the reactions that convert NH3, CO2, and the amide nitrogen of aspartate into urea.
![]() Indicate the subcellular locations of the enzymes that catalyze urea biosynthesis, and the roles of allosteric regulation and of acetylglutamate in the regulation of this process.
Indicate the subcellular locations of the enzymes that catalyze urea biosynthesis, and the roles of allosteric regulation and of acetylglutamate in the regulation of this process.
![]() Explain why metabolic defects in different enzymes of urea biosynthesis, although distinct at the molecular level, present similar clinical signs and symptoms.
Explain why metabolic defects in different enzymes of urea biosynthesis, although distinct at the molecular level, present similar clinical signs and symptoms.
![]() Describe both the classical approaches and the role of tandem mass spectrometry in screening neonates for inherited metabolic diseases.
Describe both the classical approaches and the role of tandem mass spectrometry in screening neonates for inherited metabolic diseases.
BIOMEDICAL IMPORTANCE
This chapter describes how the nitrogen of amino acids is converted to urea and the rare metabolic disorders that accompany defects in urea biosynthesis. In normal adults, nitrogen intake matches nitrogen excreted. Positive nitrogen balance, an excess of ingested over excreted nitrogen, accompanies growth and pregnancy. Negative nitrogen balance, where output exceeds intake, may follow surgery, advanced cancer, and the nutritional disorders kwashiorkor, and marasmus. Ammonia, which is highly toxic, arises in humans primarily from the α-amino nitrogen of amino acids. Tissues therefore convert ammonia to the amide nitrogen of the nontoxic amino acid glutamine. Subsequent deamination of glutamine in the liver releases ammonia, which is then converted to urea, which is not toxic. If liver function is compromised, as in cirrhosis or hepatitis, elevated blood ammonia levels generate clinical signs and symptoms. Each enzyme of the urea cycle provides examples of metabolic defects and their physiologic consequences, and the cycle as a whole serves as a molecular model for the study of human metabolic defects.
PROTEIN TURNOVER OCCURS IN ALL FORMS OF LIFE
The continuous degradation and synthesis (turnover) of cellular proteins occur in all forms of life. Each day, humans turn over 1-2% of their total body protein, principally muscle protein. High rates of protein degradation occur in tissues that are undergoing structural rearrangement, for example, uterine tissue during pregnancy, skeletal muscle in starvation, and tadpole tail tissue during metamorphosis. Approximately 75% of the amino acids liberated by protein degradation are reutilized. Excess free amino acids are, however, not stored. Those not immediately incorporated into new protein are rapidly degraded. The major portion of the carbon skeletons of the amino acids is converted to amphibolic intermediates, while in humans the amino nitrogen is converted to urea and excreted in the urine.
PROTEASES & PEPTIDASES DEGRADE PROTEINS TO AMINO ACIDS
The relative susceptibility of a protein to degradation is expressed as its half-life (t1/2), the time required to lower its concentration to half the initial value. Half-lives of liver proteins range from under 30 min to over 150 h. Typical “housekeeping” enzymes have t1/2 values of over 100 h. By contrast, key regulatory enzymes may have t1/2 values as low as 0.5-2 h. PEST sequences, regions rich in proline (P), glutamate (E), serine (S), and threonine (T), target some proteins for rapid degradation. Intracellular proteases hydrolyze internal peptide bonds. The resulting peptides are then degraded to amino acids by endopeptidases that cleave internal peptide bonds, and by aminopeptidases and carboxypeptidases that remove amino acids sequentially from the amino- and carboxyl-termini, respectively.
ATP-Independent Degradation
Degradation of blood glycoproteins (see Chapter 47) follows loss of a sialic acid moiety from the nonreducing ends of their oligosaccharide chains. Asialoglycoproteins are then internalized by liver-cell asialoglycoprotein receptors and degraded by lysosomal proteases. Extracellular, membrane-associated, and long-lived intracellular proteins are degraded in lysosomes by ATP-independent processes.
ATP and Ubiquitin-Dependent Degradation
Degradation of regulatory proteins with short half-lives and of abnormal or misfolded proteins occurs in the cytosol, and requires ATP and ubiquitin. Ubiquitin, so named because it is present in all eukaryotic cells, is a small (8.5 kDa, 76 residues) polypeptide that targets many intracellular proteins for degradation. The primary structure of ubiquitin is highly conserved. Only three of 76 residues differ between yeast and human ubiquitin. Ubiquitin molecules are attached by non-α-peptide bonds formed between the carboxyl terminal of ubiquitin and the ε-amino groups of lysyl residues in the target protein (Figure 28–1). The residue present at its amino terminal affects whether a protein is ubiquitinated. Amino terminal Met or Ser retards, whereas Asp or Arg accelerates ubiquitination. Attachment of a single ubiquitin molecule to transmembrane proteins alters their subcellular localization and targets them for degradation. Soluble proteins undergo polyubiquitination, the ligase-catalyzed attachment of four or more additional ubiquitin molecules. Subsequent degradation of ubiquitin-tagged proteins takes place in the pro-teasome, a macromolecule with multiple different subunits that also is ubiquitous in eukaryotic cells (see Chapter 46). For the discovery of ubiquitin-mediated protein degradation, Aaron Ciechanover and Avram Hershko of Israel and Irwin Rose of the United States were awarded the 2004 Nobel Prize in Chemistry. Metabolic diseases associated with defects of ubiquitination include the Angelman syndrome and the von Hippel-Lindau syndrome in which there is a defect in the ubiquitin E3 ligase. For additional aspects of protein degradation and of ubiquitination, including its role in the cell cycle, see Chapters 4 and 46.
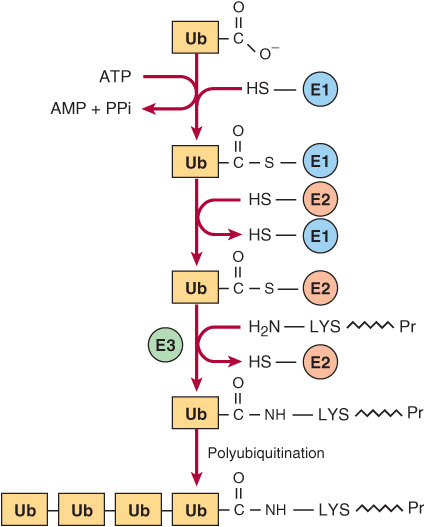
FIGURE 28–1 Reactions involved in the attachment of ubiquitin (Ub) to proteins. Three enzymes are involved. E1 is an activating enzyme, E2 a ligase, and E3 a transferase. While depicted as single entities, there are several types of E1, and over 500 types of E2. The terminal COOH of ubiquitin first forms a thioester. The coupled hydrolysis of PPi by pyrophosphatase ensures that the reaction will proceed readily. A thioester exchange reaction now transfers activated ubiquitin to E2. E3 then catalyzes the transfer of ubiquitin to the ε-amino group of a lysyl residue of the target protein. Additional rounds of ubiquitination result in subsequent polyubiquitination.
INTERORGAN EXCHANGE MAINTAINS CIRCULATING LEVELS OF AMINO ACIDS
The maintenance of steady-state concentrations of circulating plasma amino acids between meals depends on the net balance between release from endogenous protein stores and utilization by various tissues. Muscle generates over half of the total body pool of free amino acids, and liver is the site of the urea cycle enzymes necessary for disposal of excess nitrogen. Muscle and liver thus play major roles in maintaining circulating amino acid levels.
Figure 28–2 summarizes the postabsorptive state. Free amino acids, particularly alanine and glutamine, are released from muscle into the circulation. Alanine, which appears to be the vehicle of nitrogen transport in the plasma, is extracted primarily by the liver. Glutamine is extracted by the gut and the kidney, both of which convert a significant portion to alanine. Glutamine also serves as a source of ammonia for excretion by the kidney. The kidney provides a major source of serine for uptake by peripheral tissues, including liver and muscle. Branched-chain amino acids, particularly valine, are released by muscle and taken up predominantly by the brain.
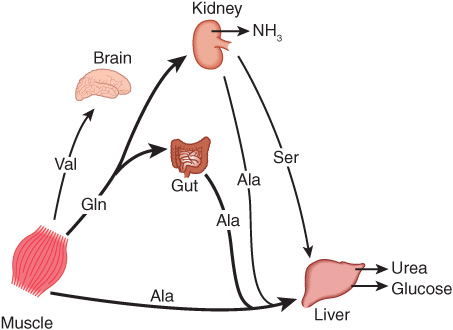
FIGURE 28–2 Interorgan amino acid exchange in normal postabsorptive humans. The key role of alanine in amino acid output from muscle and gut and uptake by the liver is shown.
Alanine is a key gluconeogenic amino acid (Figure 28–3). The rate of hepatic gluconeogenesis from alanine is far higher than from all other amino acids. The capacity of the liver for gluconeogenesis from alanine does not reach saturation until the alanine concentration reaches 20-30 times its normal physiologic level. Following a protein-rich meal, the splanchnic tissues release amino acids (Figure 28–4) while the peripheral muscles extract amino acids, in both instances predominantly branched-chain amino acids. Branched-chain amino acids thus serve a special role in nitrogen metabolism: in the fasting state, when they provide the brain with an energy source, and after feeding, when they are extracted predominantly by muscle, having been spared by the liver.
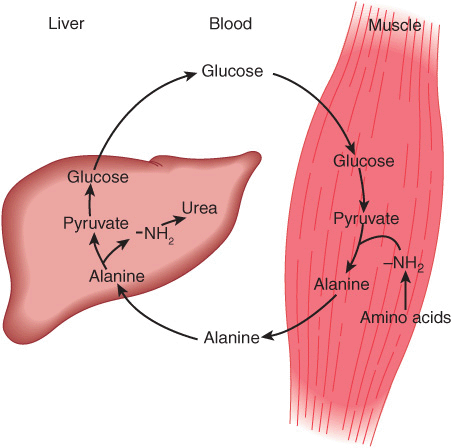
FIGURE 28–3 Theglucose-alaninecycle. Alanineissynthesized in muscle by transamination of glucose-derived pyruvate, released into the bloodstream, and taken up by the liver. In the liver, the carbon skeleton of alanine is reconverted to glucose and released into the bloodstream, where it is available for uptake by muscle and resynthesis of alanine.
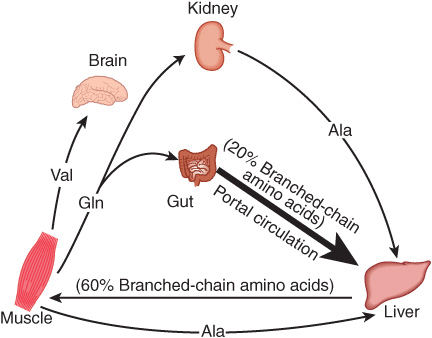
FIGURE 28–4 Summary of amino acid exchange between organs immediately after feeding.
ANIMALS CONVERT α-AMINO NITROGEN TO VARIED END PRODUCTS
Depending on their ecological niche and physiology, different animals excrete excess nitrogen as ammonia, as uric acid, or as urea. The aqueous environment of teleostean fish, which are ammonotelic (excrete ammonia), permits them to excrete water continuously to facilitate excretion of ammonia, which is highly toxic. While this approach is appropriate for an aquatic animal, birds must both conserve water and maintain low weight. Birds, which are uricotelic, address both problems by excreting nitrogen-rich uric acid (see Figure 33–11) as semisolid guano. Many land animals, including humans, are ureotelic and excrete nontoxic, highly water-soluble urea. Since urea is nontoxic to humans, high blood levels in renal disease are a consequence, not a cause, of impaired renal function.
BIOSYNTHESIS OF UREA
Urea biosynthesis occurs in four stages: (1) transamination, (2) oxidative deamination of glutamate, (3) ammonia transport, and (4) reactions of the urea cycle (Figure 28–5). The use of complementary DNA probes has shown that the expression in liver of the RNAs for all the enzymes of the urea cycle increases several fold in starvation.
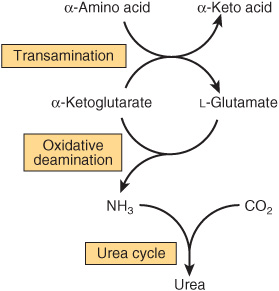
FIGURE 28–5 Overall flow of nitrogen in amino acid catabolism.
Transamination Transfers α-Amino Nitrogen to α-Ketoglutarate, Forming Glutamate
Transamination reactions interconvert pairs of α-amino acids and α-keto acids (Figure 28–6). Transamination reactions, which are freely reversible, also function in amino acid biosynthesis (see Figure 27–3). All of the common amino acids except lysine, threonine, proline, and hydroxyproline participate in transamination. Transamination is not restricted to α-amino groups. The δ-amino group of ornithine (but not the ε-amino group of lysine) readily undergoes transamination.
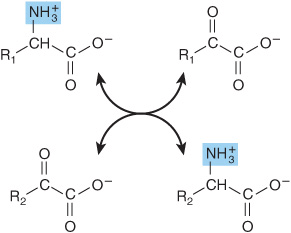
FIGURE 28–6 Transamination. The reaction is freely reversible with an equilibrium constant close to unity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


