Cardiovascular Disorders
INTRODUCTION
The cardiovascular system begins its activity when the fetus is barely a month old and is the last body system to cease activity at the end of life. This system is so vital that its activity defines the presence of life.
LIFE-GIVING TRANSPORT SYSTEM
The heart, arteries, veins, and lymphatics form the cardiovascular network that serves as the body’s transport system, bringing life-supporting oxygen and nutrients to cells, removing metabolic waste products, and carrying hormones from one part of the body to another. Often called the circulatory system, it may be divided into two branches: pulmonary circulation, in which blood picks up new oxygen and liberates the waste product carbon dioxide; and systemic circulation (including coronary circulation), in which blood carries oxygen and nutrients to all active cells while transporting waste products to the kidneys, liver, and skin for excretion.
Circulation requires normal functioning of the heart, which propels blood through the system by continuous rhythmic contractions. Located behind the sternum, the heart is a muscular organ the size of a man’s fist. It has three layers: the endocardium—the smooth inner layer; the myocardium—the thick, muscular middle layer that contracts in rhythmic beats; and the epicardium—the thin, serous membrane, or outer surface of the heart. Covering the entire heart is a saclike membrane called the pericardium, which has two layers: a visceral layer that’s in contact with the heart and a parietal, or outer, layer. To prevent irritation when the heart moves against this layer during contraction, fluid lubricates the parietal pericardium.
The heart has four chambers: two thin-walled chambers called atria and two thick-walled chambers called ventricles. The atria serve as reservoirs during ventricular contraction (systole) and as booster pumps during ventricular relaxation (diastole). The left ventricle propels blood through the systemic circulation. The right ventricle, which forces blood through the pulmonary circulation, is much thinner than the left because it meets only one sixth the resistance.
As a person’s body ages, the ventricular and aortic walls stiffen, decreasing the heart’s pumping action.
HEART VALVES
Two kinds of valves work inside the heart: atrioventricular ană semilunar. The atrioventricular valve between the right atrium and ventricle has three leaflets, or cusps, and three papillary muscles; hence, it’s called the tricuspid valve. The atrioventricular valve between the left atrium and ventricle consists of two cusps shaped like a bishop’s miter and two papillary muscles and is called the mitral valve. The tricuspid and mitral valves prevent blood backflow from the ventricles to the atria during ventricular contraction. The leaflets of both valves are attached to the ventricles’ papillary muscles by thin, fibrous bands called chordae tendineae; the leaflets separate and descend funnel-like into the ventricles during diastole and are pushed upward and together during systole to occlude the mitral and tricuspid orifices. The valves’ action isn’t entirely passive because papillary muscles contract during systole and prevent the leaflets from prolapsing into the atria during ventricular contraction.
The two semilunar valves, which resemble half-moons, prevent blood backflow from the aorta and pulmonary arteries into the ventricles when those chambers relax and fill with blood from the atria. They’re referred to as the aortic valve and pulmonic valve for their respective arteries.
In elderly people, fibrotic and sclerotic changes thicken heart valves and reduce their flexibility. These changes lead to rigidity and incomplete closure of the valves, which may result in systolic or diastolic murmurs
THE CARDIAC CYCLE
Diastole is the phase of ventricular relaxation and filing. As diastole begins, ventricular pressure falls below arterial pressure, and the aortic and pulmonic valves close. As ventricular pressure continues to fall below atrial pressure, the mitral and tricuspid valves open, and blood flows rapidly into the ventricles. Atrial contraction then increases the volume of ventricular filing by pumping 15% to 25% more blood into the ventricles. When systole begins, the ventricular muscle contracts, raising ventricular pressure above atrial pressure and closing the mitral and tricuspid valves. When ventricular pressure finally becomes greater than that in the aorta and pulmonary artery, the aortic and pulmonic valves open, and the ventricles eject blood. Ventricular pressure continues to rise as blood is expelled from the heart. As systole ends, the ventricles relax and stop ejecting blood, and ventricular pressure falls, closing both valves.
S1 (the first heart sound) is heard as the ventricles contract and the atrioventricular valves close. S1 is loudest at the heart’s apex, over the
mitral area. S2 (the second heart sound), which is normally rapid and sharp, occurs when the aortic and pulmonic valves close. S2 is loudest at the heart’s base (second intercostal space on both sides of the sternum).
mitral area. S2 (the second heart sound), which is normally rapid and sharp, occurs when the aortic and pulmonic valves close. S2 is loudest at the heart’s base (second intercostal space on both sides of the sternum).
Normally, with inspiration, a split S2 will be auscultated. With expiration, the split S2 sounds will occur closer together or the S2 may become a single sound. However, a fixed split S2 will be heard if the patient has a right bundle-branch block.
Ventricular distention during diastole, which can occur in heart failure, creates low-frequency vibrations that may be heard as a third heart sound (S3), or ventricular gallop. An atrial gallop (S4) may appear at the end of diastole, just before S1, if atrial filling is forced into a ventricle that has become less compliant or overdistended or has a decreased ability to contract. A pressure rise and ventricular vibrations cause this sound.
CARDIAC CONDUCTION
The heart’s conduction system is composed of specialized cells capable of generating and conducting rhythmic electrical impulses to stimulate heart contraction. This system includes the sinoatrial (SA) node, the atrioventricular (AV) junction, the bundle of His and its bundle branches, and the ventricular conduction tissue and Purkinje fibers.
Normally, the SA node controls the heart rate and rhythm at 60 to 100 beats/minute. Because the SA node has the lowest resting potential, it’s the heart’s pacemaker. If it defaults, another part of the system takes over. The AV junction may emerge at 40 to 60 beats/minute; the bundle of His and bundle branches at 30 to 40 beats/minute; and ventricular conduction tissue at 20 to 30 beats/minute.
As the myocardium of the aging heart becomes more irritable, extrasystoles may occur along with sinus arrhythmias and sinus bradycardias. In addition, increased fibrous tissue infiltrates the SA nodes andinternodal atrial tracts, which may cause atrial fibrillation and flutter.
CARDIAC OUTPUT
Cardiac output—the amount of blood pumped by the left ventricle into the aorta each minute—is calculated by multiplying the stroke volume (the amount of blood the left ventricle ejects during each contraction) by the heart rate (number of beats/minute). When cellular demands increase, stroke volume or heart rate must increase.
Many factors affect the heart rate, including exercise, pregnancy, and stress. When the sympathetic nervous system releases norepinephrine, the heart rate increases; when the parasympathetic system releases acetylcholine, it slows. As a person ages, the heart rate takes longer to normalize after exercise.
Stroke volume depends on the ventricular blood volume and pressure at the end of diastole (preload), resistance to ejection (afterload), and the myocardium’s contractile strength (inotropy). Changes in preload, afterload, or inotropic state can alter the stroke volume.
Exercise cardiac output declines slightly with age. A decrease in maximum heart rate and contractility may cause this change.
CIRCULATION AND PULSES
Blood circulates through three types of vessels: arteries, veins, and capillaries. The sturdy, pliable walls of the arteries adjust to the volume of blood leaving the heart. The major artery branching out of the left ventricle is the aorta. Its segments and subbranches ultimately divide into minute, thin-walled (one-cell thick) capillaries. Capillaries pass the blood to the veins, which return it to the heart. In the veins, valves prevent blood backflow.
Aging contributes to arterial and venous insufficiency as the strength and elasticity of blood vessels decrease.
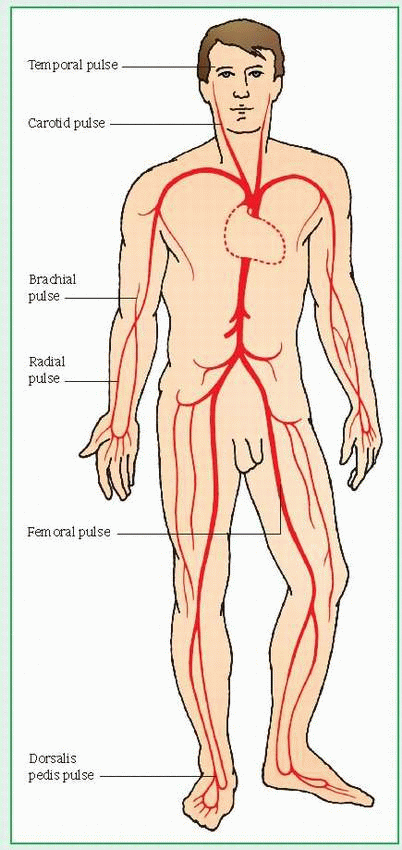
Pulses are felt best wherever an artery runs near the skin and over a hard structure. (See Pulse points.) Easily found pulses are:
radial artery—anterolateral aspect of the wrist
temporal artery—in front of the ear, above and lateral to the eye
common carotid artery—neck (side)
femoral artery—groin
The lymphatic system also plays a role in the cardiovascular network. Originating in tissue spaces, the lymphatic system drains fluid and other plasma components that build up in extravascular spaces and reroutes them back to the circulatory system as lymph, a plasmalike fluid. Lymphatics also extract bacteria and foreign bodies.
CARDIOVASCULAR ASSESSMENT
Physical assessment provides vital information about cardiovascular status.
Check for underlying cardiovascular disorders, such as central cyanosis (impaired gas exchange), edema (heart failure or valvular disease), and clubbing (congenital cardiovascular disease).
Palpate the peripheral pulses bilaterally and evaluate their rate, equality, and quality on a
scale of 0 (absent) to +4 (bounding). (See Pulse amplitude scale.)
Pulse points
Peripheral pulse rhythm should correspond exactly to the auscultatory heart rhythm. The pulse’s character may offer useful information. For example, pulsus alternans, a strong beat followed by a weak one, can mean myocardial weakness. A water-hammer (or Corrigan’s) pulse, a forceful bounding pulse best felt in the carotid arteries or in the forearm, accompanies increased pulse pressure—commonly with capillary pulsations of the fingernails (Quincke’s sign). This pulse usually indicates patent ductus arteriosus or aortic insufficiency. Pulsus bisferiens, a double peripheral pulse for every apical beat, can signal aortic stenosis, hyperthyroidism, or some other disease. Pulsus bigeminus is a coupled rhythm; you feel its beat in pairs. Pulsus paradoxus is exaggerated waxing and waning of the arterial pressure (>15 mm Hg decrease in systolic blood pressure during inspiration).
 |
Inspect the carotid arteries for equal appearance. Auscultate for bruits; then palpate the arteries individually, one side at a time, for thrills (fine vibrations due to irregular blood flow).
Check for pulsations in the jugular veins (more easily seen than felt). Watch for jugular vein distention—a possible sign of right-sided heart failure, valvular stenosis, cardiac tamponade, or pulmonary embolism. Take blood pressure readings in both arms while the patient is lying, sitting, and standing.
Palpate the precordium for any abnormal pulsations, such as lifts, heaves, or thrills. Use the palms (at the base of the fingertips) or the fingertips. The normal apex will be felt as a light tap and extends over 1 “ (2.5 cm) or less.
Systematically auscultate the anterior chest wall for each of the four heart sounds in the aortic area (second intercostal space at the right sternal border), pulmonic area (second intercostal space at the left sternal border), right ventricular area (lower half of the left sternal border), and mitral area (fifth intercostal space at the midclavicular line). However, don’t limit your auscultation to these four areas. Valvular sounds may be heard all over the precordium. Therefore, inch your stethoscope in a Z pattern, from the base of the heart across and down and then over to the apex, or start at the apex and work your way up. For low-pitched sounds, use the bell of the stethoscope; for high-pitched sounds, the diaphragm. Carefully inspect each area for pulsations, and palpate for thrills. Check the location of apical pulsation for deviations in normal size (3/8” to3/4” [1 to 2 cm]) and position (in the mitral area)—possible signs of left ventricular hypertrophy, left-sided valvular disease, or right ventricular disease.
Listen for the vibrating sound of turbulent blood flow through a stenotic or incompetent valve. Time the murmur to determine where
it occurs in the cardiac cycle—between S1 and S2 (systolic), between S2 and the following S1 (diastolic), or throughout systole (holosystolic). Finally, listen for the scratching or squeaking of a pericardial friction rub.
SPECIAL CARDIOVASCULAR TESTS
Electrocardiography (ECG) measures electrical activity by recording currents transmitted by the heart. It can detect ischemia, injury, necrosis, bundle-branch blocks, fascicular blocks, conduction delay, chamber enlargement, and arrhythmias. In Holter monitoring, a tape recording tracks as many as 100,000 cardiac cycles over a 12- or 24-hour period. This test may be used to assess the effectiveness of antiarrhythmic drugs or to evaluate arrhythmia symptoms. A signal-averaged ECG will identify afterpotentials, which are associated with a risk of ventricular arrhythmias. (See Positioning chest electrodes.)
Chest X-rays may reveal cardiac enlargement and aortic dilation. They also assess pulmonary circulation. When pulmonary venous and arterial pressures rise, characteristic changes appear, such as dilation of the pulmonary venous shadows. When pulmonary venous pressure exceeds oncotic pressure of the blood, capillary fluid leaks into lung tissues, causing pulmonary edema. This fluid may settle in the alveoli, producing a butterfly pattern, or the lungs may appear cloudy or hazy; in the interlobular septa, sharp linear densities (Kerley’s lines) may appear.
Pulse amplitude scale
To record your patient’s pulse amplitude, use this standard scale:
0: Pulse isn’t palpable. + 1: Pulse is thready, weak, difficult to find, may fade in and out, and disappears easily with pressure.
+2: Pulse is constant but not strong; light pressure must be applied or pulse will disappear.
+3: Pulse considered normal. Is easily palpable, doesn’t disappear with pressure. +4: Pulse is strong, bounding, and doesn’t disappear with pressure.
Exercise testing using a bicycle ergometer or treadmill determines the heart’s response to physical stress. This test measures blood pressure and ECG changes during increasingly rigorous exercises. Myocardial ischemia, abnormal blood pressure response, or arrhythmias indicate the circulatory system’s failure to adapt to exercise.
Cardiac catheterization evaluates chest pain, the need for coronary artery surgery or angioplasty, congenital heart defects, and valvular heart disease and determines the extent of heart failure. Right-sided catheterization involves threading a pulmonary artery thermodilution catheter, which can measure cardiac output, through a vein into the right side of the heart, pulmonary artery, and its branches in the lungs to measure right atrial, right ventricular, pulmonary artery, and pulmonary artery wedge pressures. Left-sided catheterization entails retrograde catheterization of the left ventricle or transseptal catheterization of the left atrium. Ventriculography during left-sided catheterization involves injecting radiopaque dye into the left ventricle to measure ejection fraction and to disclose abnormal heart wall motion or mitral valve incompetence.
In coronary arteriography, radiopaque material injected into coronary arteries allows cineangiographic visualization of coronary arterial narrowing or occlusion.
Positioning chest electrodes
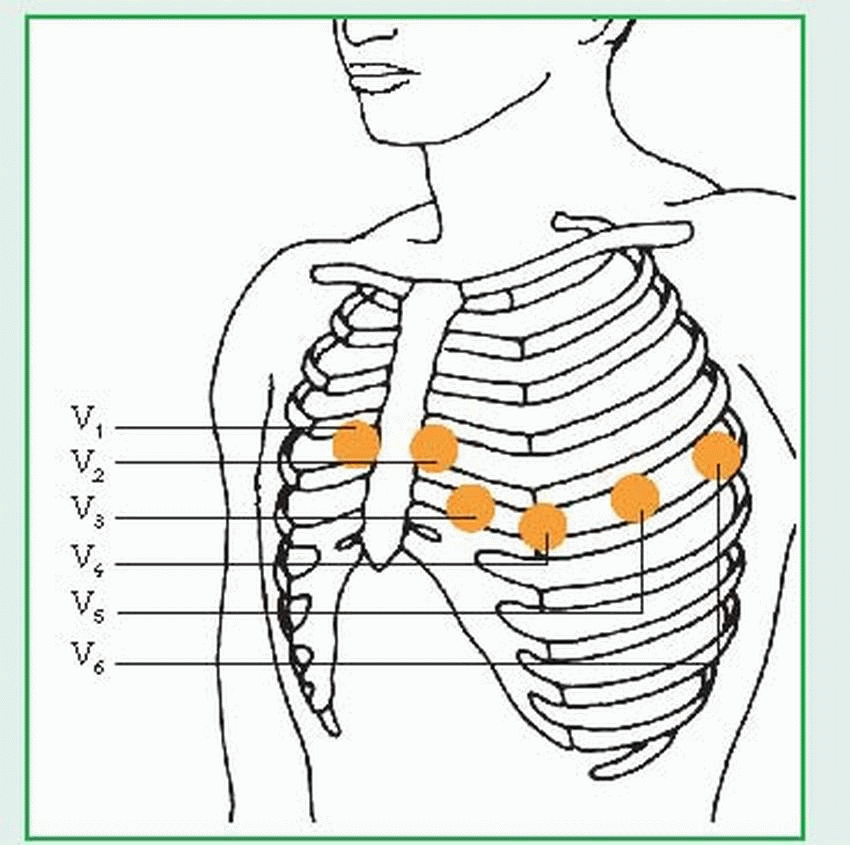
To record the 12-lead electrocardiogram, place electrodes on the patient’s arms and legs (with the ground lead on the patient’s right leg). The three standard limb leads (I, II, III) and the three augmented leads (aVR, aVL, aVF) are recorded using these electrodes. To record the precordial (chest) leads, place the electrodes as follows:
V1 —fourth intercostal space (ICS), right sternal border
V2—fourth ICS, left sternal border
V3—midway between V2 and V4
V4—fifth ICS, left midclavicular line
V5—fifth ICS, left anterior axillary line
V6—fifth ICS, left midaxillary line
 |
Digital subtraction angiography evaluates the coronary arteries through the use of X-ray studies in which images of bone and soft tissue are digitally subtracted by computer. Time-based color enhancement shows blood flow in nearby areas.
Echocardiography uses echoes from pulsed high-frequency sound waves (ultrasound) to evaluate cardiac structures. M-mode echocardiography, in which a single, stationary ultrasound beam strikes the heart, produces a vertical view of cardiac structures. Two-dimensional echocardiography (most common), in which an ultrasound beam rapidly sweeps through an arc, produces a cross-sectional or fan-shaped view of cardiac structures. Both M-mode and two-dimensional echocardiography may use contrast agents for image enhancement. Doppler echocardiography records blood flow within the cardiovascular system. Color Doppler echocardiography shows the direction of blood flow, which provides information about the degree of valvular insufficiency. Transesophageal echocardiography combines ultrasound with endoscopy to better view the heart’s structures. This procedure allows images to be taken from the heart’s posterior aspect.
Echocardiography provides information about valve leaflets, size and dimensions of heart chambers, and thickness and motion of the septum and the ventricular walls. It can also reveal intracardiac masses, detect pericardial effusion, diagnose hypertrophic cardiomyopathy, and estimate cardiac output and ejection fraction. This test can also evaluate possible aortic dissection when it involves the ascending aorta.
In multiple-gated acquisition scanning, a radioactive isotope in the intravascular compartment allows measurement of stroke volume, wall motion, and ventricular ejection fraction. Myocardial imaging uses the radioactive agent thallium-201 or Tc-99m sestamibi to detect abnormalities in myocardial perfusion. This agent concentrates in normally perfused areas of the myocardium but not in ischemic areas (“cold spots”), which may be permanent (scar tissue) or temporary (from transient ischemia). These tests can be done as exercise studies or can be combined with drugs, such as adenosine or dipyridamole, in patients unable to exercise.
Acute infarct imaging documents muscle viability (not perfusion) through the use of technetium-labeled pyrophosphate. Unlike thallium, technetium accumulates only in irreversibly damaged myocardial tissue. Areas of necrosis appear as “hot spots” and can be detected only during an acute myocardial infarction (MI). This test determines the size and location of an infarction but can produce false results.
Blood tests
Cardiac enzymes (cellular proteins released into blood after cell membrane injury) confirm acute MI or severe cardiac trauma. All cardiac enzymes—creatine kinase (CK), lactate dehydrogenase, and aspartate aminotransferase, for example—are also found in other cells. Fractionation of enzymes can determine the source of damaged cells. For example, three fractions of CK are isolated, one of which (an isoenzyme called CK-MB) is found only in cardiac cells. CK-MB in the blood indicates injury to myocardial cells.
Measurement of a cardiac protein called troponin is the most precise way to determine if a patient has experienced an MI. Some 6 hours after an MI, a blood test can detect two forms of troponin: T and I. Troponin T levels peak about 2 days after an MI and return to normal about 16 days later. Troponin I levels reach their peak in less than 1 day after an MI and return to normal in about 7 days.
Peripheral arteriography consists of a fluoroscopic X-ray after arterial injection of a contrast medium. Similarly, phlebography defines the venous system after injection of a contrast medium into a vein. Impedance plethysmography evaluates the venous system to detect pressure changes transmitted to lower leg veins.
Doppler ultrasonography evaluates the peripheral vascular system and assesses peripheral artery disease when combined with sequential systolic blood pressure readings.
Endomyocardial biopsy can detect cardiomyopathy, infiltrative myocardial diseases, and transplant rejection.
Electrophysiologic studies help diagnose conduction system disease and serious arrhythmias. Electronic induction and termination of arrhythmias aid drug selection. Endocardial mapping detects an arrhythmia’s focus using a finger electrode. Epicardial mapping uses a computer and a fabric sock with electrodes that’s slipped over the heart to detect arrhythmias.
Magnetic resonance imaging can investigate cardiac structure and function. Positron emission tomography and magnetic resonance spectroscopy are used to assess myocardial metabolism.
Electron beam computed tomography, also known as ultrafast computed tomography, is used to detect microcalcifications in the coronary arteries. This test is useful for identifying early coronary artery disease.
MANAGING CARDIOVASCULAR DISEASE
Patients with cardiovascular disease pose a tremendous challenge. Their sheer numbers alone compel a thorough understanding of cardiovascular anatomy, physiology, and pathophysiology. Anticipate a high anxiety level in cardiac patients, and provide support and reassurance, especially during procedures such as cardiac catheterization.
Cardiac rehabilitation programs are widely prescribed and offer education and support along with exercise instruction. Rehabilitation programs begin in health care facilities and continue on an outpatient basis. Helping the patient resume a satisfying lifestyle requires planning and comprehensive teaching. Inform the patient about health care facilities and organizations that offer cardiac rehabilitation programs.
CONGENITAL ACYANOTIC DEFECTS
Ventricular septal defect
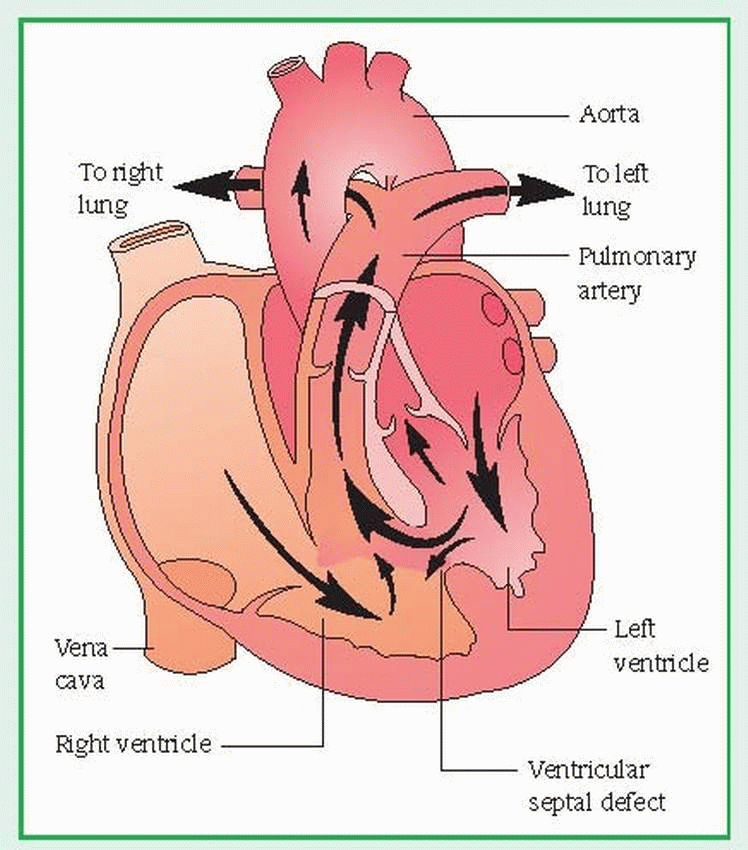
In ventricular septal defect (VSD), the most common congenital heart disorder, an opening in the septum between the ventricles allows blood to shunt between the left and right ventricles. This disease accounts for up to 30% of all congenital heart defects. The prognosis is good for defects that close spontaneously or are correctable surgically but poor for untreated defects, which are sometimes fatal by age 1, usually from secondary complications. (See Understanding ventricular septal defect.)
CAUSES AND INCIDENCE
In neonates with VSD, the ventricular septum fails to close completely by the eighth week of gestation, as it would normally. VSD occurs in some neonates with fetal alcohol syndrome, but a causal relationship hasn’t been established. Although most children with congenital heart defects are otherwise normal, in some, VSD coexists with additional birth defects, especially Down syndrome and other autosomal trisomies, renal anomalies, and such cardiac defects as patent ductus arteriosus and coarctation of the aorta. VSDs are located in the membranous or muscular portion of the ventricular septum and vary in size. Some defects close spontaneously; in other defects, the entire septum is absent, creating a single ventricle.
VSD isn’t readily apparent at birth, because right and left ventricular pressures are about equal, so blood doesn’t shunt through the defect. As the pulmonary vasculature gradually
relaxes, 4 to 8 weeks after birth, right ventricular pressure decreases, allowing blood to shunt from the left to the right ventricle.
relaxes, 4 to 8 weeks after birth, right ventricular pressure decreases, allowing blood to shunt from the left to the right ventricle.
Understanding ventricular septal defect
A ventricular septal defect (VSD), the most common type of congenital disorder, is an abnormal opening between the right and left ventricles that allows blood to shunt between them. Not always readily apparent at birth, the defect can be small and may close spontaneously. The septum may be entirely absent, resulting in a single ventricle. A large, untreated defect can cause right ventricular hypertrophy, pulmonary hypertension, and heart failure. VSD is classified as an increased pulmonary blood flow defect.
 |
Less than 1% of neonates are born with VSD. In 80% to 90% of neonates who are born with this disorder, the hole is small and will usually close spontaneously. In the remaining 10% to 20% of neonates, surgery is needed to close the hole.
COMPLICATIONS
Right arterial and ventricular hypertrophy
Heart failure
Pulmonary hypertension
SIGNS AND SYMPTOMS
Clinical features of VSD vary with the defect’s size, the shunting’s effect on the pulmonary vasculature, and the infant’s age. In a small VSD, shunting is minimal, and pulmonary artery pressure and heart size remain normal. Such defects may eventually close spontaneously without ever causing symptoms.
Initially, large VSD shunts cause left atrial and left ventricular hypertrophy. Later, an uncorrected VSD will cause right ventricular hypertrophy due to increasing pulmonary vascular resistance. Eventually, biventricular heart failure and cyanosis (from reversal of shunt direction) occur. Resulting cardiac hypertrophy may make the anterior chest wall prominent. A large VSD increases the risk of pneumonia.
Infants with large VSDs are thin and small and gain weight slowly. They may develop heart failure with dusky skin; liver, heart, and spleen enlargement because of systemic venous congestion; diaphoresis; feeding difficulties; rapid, grunting respirations; and increased heart rate. They may also develop severe pulmonary hypertension. Fixed pulmonary hypertension may occur much later in life with right-to-left shunt (Eisenmenger’s syndrome), causing cyanosis and clubbing of the nail beds.
The typical murmur associated with a VSD is blowing or rumbling and varies in frequency. In the neonate, a moderately loud early systolic murmur may be heard along the lower left sternal border. About the second or third day after birth, the murmur may become louder and longer. In infants, the murmur may be loudest near the heart’s base and may suggest pulmonary stenosis. A small VSD may produce a functional murmur or a characteristic loud, harsh systolic murmur. Larger VSDs produce audible murmurs (at least a grade 3 pansystolic), loudest at the fourth intercostal space, usually with a thrill;
however, a large VSD with minimal pressure gradient may have no audible murmur. In addition, the pulmonic component of S2 sounds loud and is widely split. Palpation reveals displacement of the point of maximal impulse to the left. When fixed pulmonary hypertension is present, a diastolic murmur may be audible on auscultation, the systolic murmur becomes quieter, and S2 is greatly accentuated.
however, a large VSD with minimal pressure gradient may have no audible murmur. In addition, the pulmonic component of S2 sounds loud and is widely split. Palpation reveals displacement of the point of maximal impulse to the left. When fixed pulmonary hypertension is present, a diastolic murmur may be audible on auscultation, the systolic murmur becomes quieter, and S2 is greatly accentuated.
DIAGNOSIS
Diagnostic findings include:
Chest X-ray is normal in small defects; in large VSDs, it shows cardiomegaly, left atrial and left ventricular enlargement, and prominent pulmonary vascular markings.
Electrocardiography (ECG) is normal in children with small VSDs; in large VSDs, it shows left and right ventricular hypertrophy, suggesting pulmonary hypertension.
Echocardiography may detect a large VSD and its location in the septum, estimate the size of a left-to-right shunt, suggest pulmonary hypertension, and identify associated lesions and complications.
Cardiac catheterization determines the VSD’s size and exact location, calculates the degree of shunting by comparing the blood oxygen saturation in each ventricle, determines the extent of pulmonary hypertension, and detects associated defects.
TREATMENT
In mild cases, no treatment is needed, although the infant should be closely followed to make sure that the hole closes properly as he grows. Large defects usually require early surgical correction before heart failure and irreversible pulmonary vascular disease develop.
For small defects, surgery consists of simple suture closure. Moderate to large defects require insertion of a patch graft, using cardiopulmonary bypass. In patients with heart failure, digoxin and diuretics may be prescribed to control symptoms. In patients who develop increased pulmonary resistance and irreversible pulmonary vascular changes that produce a reversible right-to-left shunt (Eisenmenger’s syndrome), a heart-lung transplant may be required.
If the child has other defects and will benefit from delaying surgery, pulmonary artery banding normalizes pressures and flow distal to the band and prevents pulmonary vascular disease, allowing postponement of surgery. (Pulmonary artery banding is done only when the child has other complications.) A rare complication of VSD repair is complete heart block from interference with the bundle of His during surgery. (Heart block may require temporary or permanent pacemaker implantation.)
Before surgery, treatment consists of
digoxin, sodium restriction, and diuretics to prevent heart failure
careful monitoring by physical examination, X-ray, and ECG to detect increased pulmonary hypertension, which indicates a need for early surgery
measures to prevent infection (prophylactic antibiotics, e.g., to prevent infective endocarditis)
Generally, postoperative treatment includes a brief period of mechanical ventilation. The patient will need analgesics and may also require diuretics to increase urine output, continuous infusions of nitroprusside or adrenergic agents to regulate blood pressure and cardiac output and, in rare cases, a temporary pacemaker.
SPECIAL CONSIDERATIONS
Although the parents of an infant with VSD often suspect something is wrong with their child before diagnosis, they need psychological support to help them accept the reality of a serious cardiac disorder. Because surgery may take place months after diagnosis, parent teaching is vital to prevent complications until the child is scheduled for surgery or the defect closes. Thorough explanations of all tests are also essential.
Instruct parents to watch for signs of heart failure, such as poor feeding, sweating, and heavy breathing.
If the child is receiving digoxin or other medications, tell the parents how to give it and how to recognize adverse effects. Caution them to keep medications out of the reach of all children.
Teach parents to recognize and report early signs of infection and to avoid exposing the child to people with obvious infections.
Encourage parents to let the child engage in normal activities.
Tell parents to follow up with their pediatrician. Also tell them that child life therapy may be appropriate if their child displays delayed growth and development or failure to thrive.
Stress the importance of prophylactic antibiotics before and after surgery.
After surgery to correct VSD:
Monitor vital signs and intake and output. Maintain the infant’s body temperature with an overbed warmer. Give catecholamines, nitroprusside, and diuretics, as ordered; analgesics as needed.
Monitor central venous pressure, intra-arterial blood pressure, and left atrial or pulmonary
artery pressure readings. Assess heart rate and rhythm for signs of conduction block.
Check oxygenation, particularly in a child who requires mechanical ventilation. Suction to maintain a patent airway and to prevent atelectasis and pneumonia, as needed.
Monitor pacemaker effectiveness if needed. Watch for signs of failure, such as bradycardia and hypotension.
Reassure parents and allow them to participate in their child’s care.
Atrial septal defect
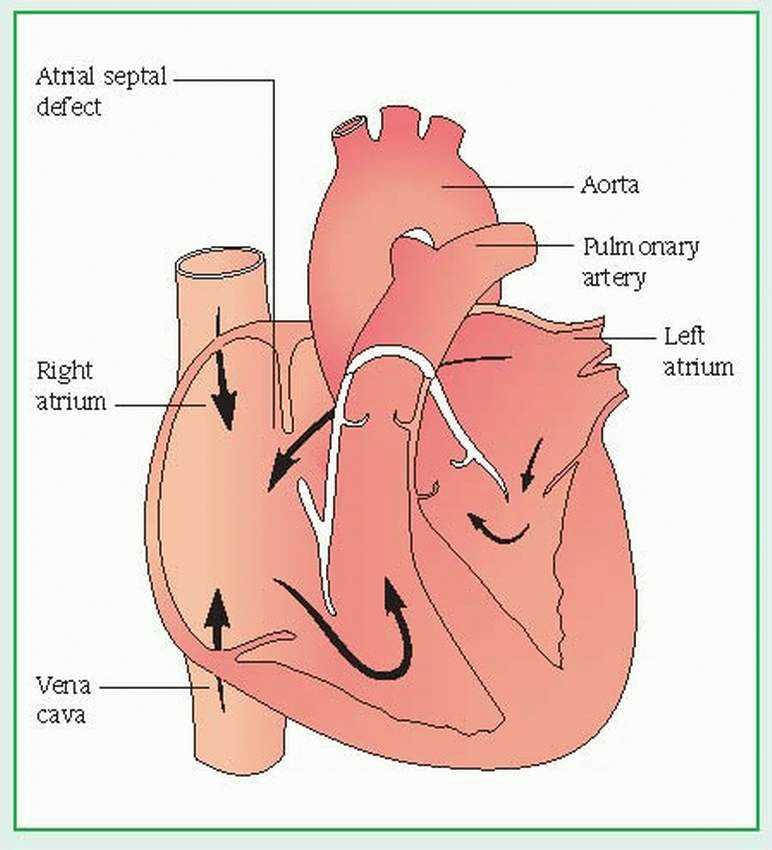
In an atrial septal defect (ASD), an opening between the left and right atria allows shunting of blood between the chambers. Ostium secundum defect (most common) occurs in the region of the fossa ovalis and occasionally extends inferiorly, close to the vena cava; sinus venosus defect occurs in the superior-posterior portion of the atrial septum, sometimes extending into the vena cava, and is almost always associated with abnormal drainage of pulmonary veins into the right atrium; ostium primum defect occurs in the inferior portion of the septum primum and is usually associated with atrioventricular valve abnormalities (cleft mitral valve) and conduction defects.
ASD accounts for about 10% of congenital heart defects and appears almost twice as often in females as in males, with a strong familial tendency. Although ASD is usually a benign defect during infancy and childhood, delayed development of symptoms and complications makes it one of the most common congenital heart defects diagnosed in adults. The prognosis is excellent in asymptomatic patients but poor in those with cyanosis caused by large, untreated defects. (See Understanding atrial septal defect.)
CAUSES AND INCIDENCE
The cause of ASD is unknown. In this condition, blood shunts from left to right because left atrial pressure normally is slightly higher than right atrial pressure; this pressure difference forces large amounts of blood through a defect. The left-to-right shunt results in right heart volume overload, affecting the right atrium, right ventricle, and pulmonary arteries. Eventually, the right atrium enlarges, and the right ventricle dilates to accommodate the increased blood volume. If pulmonary artery hypertension develops because of the shunt (rare in children), increased pulmonary vascular resistance and right ventricular hypertrophy will follow. In some adult patients, irreversible (fixed) pulmonary artery hypertension causes reversal of the shunt direction, which results in unoxygenated blood entering the systemic circulation, causing cyanosis.
ASD is present in 4 of every 100,000 people. Symptoms usually develop before age 30. When no other congenital defect exists, the patient— especially if a child—may be asymptomatic.
COMPLICATIONS
Unoxygenated blood in systemic circulation
Right and left ventricular hypertrophy
Atrial arrhythmias
Heart failure
Emboli
SIGNS AND SYMPTOMS
ASD commonly goes undetected in preschoolers; such children may complain about feeling tired only after extreme exertion and may have frequent respiratory tract infections but otherwise appear normal and healthy. However, children with large shunts may show growth retardation. Children with ASD seldom develop heart failure, pulmonary hypertension, infective endocarditis, or other complications. However, as adults, they usually manifest pronounced symptoms, such as fatigability and dyspnea on exertion, frequently to the point of severe limitation of activity (especially after age 40).
In children, auscultation reveals an early to midsystolic murmur, superficial in quality, heard at the second or third left intercostal space. In patients with large shunts (resulting from increased tricuspid valve flow), a low-pitched diastolic murmur is heard at the lower left sternal border, which becomes more pronounced on inspiration. Although the murmur’s intensity is a rough indicator of the size of the left-toright shunt, its low pitch sometimes makes it difficult to hear and, if the pressure gradient is relatively low, a murmur may not be detectable. Other signs include a fixed, widely split S2, caused by delayed closure of the pulmonic valve, and a systolic click or late systolic murmur at the apex, resulting from mitral valve prolapse, which occasionally affects older children with ASD.
In older patients with large, uncorrected defects and fixed pulmonary artery hypertension, auscultation reveals an accentuated S2. A pulmonary ejection click and an audible S4 may also be present. Clubbing and cyanosis become evident; syncope and hemoptysis may occur with severe pulmonary vascular disease.
Understanding atrial septal defect
An atrial septal defect (ASD) is an abnormal opening between the left and right atria. A small opening may cause few symptoms. However, if the opening is large, higher pressure in the left atrium can shunt large amounts of blood into the right atrium, which can result in right heart volume overload, right atrial and ventricular enlargement, and pulmonary hypertension. ASD is classified as an increased pulmonary blood flow defect and is one of the most common congenital heart defects.
 |
DIAGNOSIS
A history of increasing fatigue and characteristic physical features suggest ASD. The following findings confirm it:
Chest X-ray shows an enlarged right atrium and right ventricle, a prominent pulmonary artery, and increased pulmonary vascular markings.
Electrocardiography may be normal but usually shows right axis deviation, prolonged PR interval, varying degrees of right bundlebranch block, right ventricular hypertrophy, atrial fibrillation (particularly in severe cases after age 30) and, in ostium primum defect, left axis deviation.
Echocardiography measures right ventricular enlargement, may locate the defect, and shows volume overload in the right side of the heart. (Other causes of right ventricular enlargement must be ruled out.)
Two-dimensional echocardiography with color Doppler flow, contrast echocardiography, or both have supplanted cardiac catheterization as the con firming tests for ASD. Cardiac catheterization is used if inconsistencies exist in the clinical data or if significant pulmonary hypertension is suspected.
TREATMENT
Operative repair is advised for all patients with uncomplicated ASD with evidence of significant left-to-right shunting. Ideally, this is performed when the patient is between ages 2 and 4. Operative treatment shouldn’t be performed on patients with small defects and trivial left-to-right shunts. Because ASD seldom produces complications in infants and toddlers, surgery can be delayed until they reach preschool or early school age. A large defect may need immediate surgical closure with sutures or a patch graft.
Physicians have developed a new procedure, referred to as catheter closure or transcatheter closure of the atrial septal defect, that uses wires or catheters to close ASD without surgery. In this procedure, the surgeon makes a tiny incision in the groin to introduce the catheters. Then, he advances the catheters into the heart and places the closure device across the ASD. This procedure may not be applicable to all patients.
SPECIAL CONSIDERATIONS
Before cardiac catheterization, explain pretest and posttest procedures to the child and her parents. If possible, use drawings or other visual aids to explain it to the child.
As needed, teach the patient about prophylactic antibiotics to prevent infective endocarditis. (They may be administered before dental or other invasive procedures.)
If surgery is scheduled, teach the child and her parents about the intensive care unit and introduce them to the staff. Show parents where they can wait during the operation. Explain postoperative procedures, tubes, dressings, and monitoring equipment.
After surgery, closely monitor the patient’s vital signs, central venous and intra-arterial pressures, and intake and output. Watch for atrial arrhythmias, which may remain uncorrected.
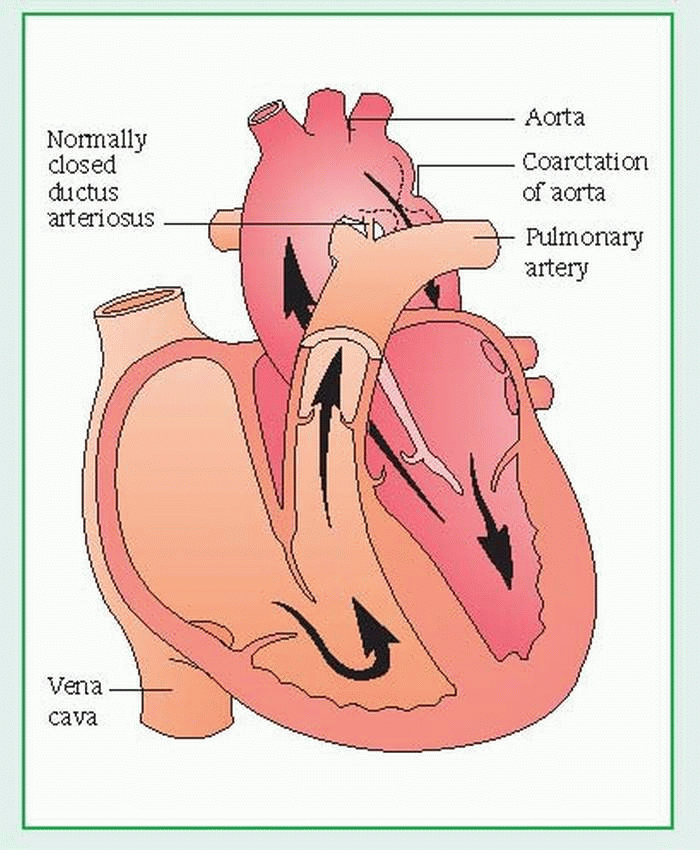
Coarctation of the aorta
Coarctation is a narrowing of the aorta, usually just below the left subclavian artery, near the site where the ligamentum arteriosum (the remnant of the ductus arteriosus, a fetal blood vessel) joins the pulmonary artery to the aorta. Coarctation may occur with aortic valve stenosis (usually of a bicuspid aortic valve) and with severe cases of hypoplasia of the aortic arch, patent ductus arteriosus, and ventricular septal defect. Generally, the prognosis for coarctation of the aorta depends on the severity of associated cardiac anomalies; the prognosis for isolated coarctation is good if corrective surgery is performed before this condition induces severe systemic hypertension or degenerative changes in the aorta. (See Understanding coarctation of the aorta.)
CAUSES AND INCIDENCE
Coarctation of the aorta may develop as a result of spasm and constriction of the smooth muscle in the ductus arteriosus as it closes. Possibly, this contractile tissue extends into the aortic wall, causing narrowing. The obstructive process causes hypertension in the aortic branches above the constriction (arteries that supply the arms, neck, and head) and diminished pressure in the vessels below the constriction.
Restricted blood flow through the narrowed aorta increases the pressure load on the left ventricle and causes dilation of the proximal aorta and ventricular hypertrophy. Untreated, this condition may lead to leftsided heart failure and, rarely, to cerebral hemorrhage and aortic rupture. If ventricular septal defect accompanies coarctation, blood shunts left to right, straining the right side of the heart. This leads to pulmonary hypertension and, eventually, right-sided heart hypertrophy and failure.
Coarctation of the aorta occurs in 1 of every 10,000 people and is usually diagnosed in children or adults younger than age 40. It accounts for about 7% of all congenital heart defects in children and is twice as common in males as in females. When it occurs in females, it’s commonly associated with Turner’s syndrome, a chromosomal disorder that causes ovarian dysgenesis.
COMPLICATIONS
Infective endocarditis
Pulmonary hypertension
Right ventricular hypertrophy
Right-sided heart failure
SIGNS AND SYMPTOMS
Clinical features vary with age. During the first year of life, when aortic coarctation may cause heart failure, the infant displays tachypnea, dyspnea, pulmonary edema, pallor, tachycardia, failure to thrive, cardiomegaly, and hepatomegaly. In most cases, heart sounds are normal unless a coexisting cardiac defect is present. Femoral pulses are absent or diminished.
If coarctation is asymptomatic in infancy, it usually remains so throughout adolescence, as collateral circulation develops to bypass the narrowed segment. During adolescence, this defect may produce dyspnea, claudication, headaches, epistaxis, and hypertension in the upper extremities despite collateral circulation. It commonly causes resting systolic hypertension and wide pulse pressure; high diastolic pressure readings are the same in both the arms and legs. Coarctation may also produce a visible aortic pulsation in the suprasternal notch, a continuous systolic murmur, an accentuated S2, and an S4.
DIAGNOSIS
The cardinal signs of coarctation of the aorta are resting systolic hypertension, absent or diminished femoral pulses, and wide pulse pressure
The following tests support this diagnosis:
Chest X-ray may demonstrate left ventricular hypertrophy, heart failure, a wide ascending and descending aorta, and notching of the undersurfaces of the ribs, due to extensive collateral circulation.
Electrocardiography may eventually reveal left ventricular hypertrophy.
Understanding coarctation of the aorta
Coarctation is a narrowing of the aorta, usually just below the left subclavian artery, near the site where the ligamentum arteriosum joins the pulmonary artery to the aorta. It can result from spasm and constriction of the smooth muscle in the ductus arteriosus as it closes. Restricted blood flow through the narrow aorta increases the pressure load on the left ventricle, resulting in dilation of the proximal aorta, left ventricular hypertrophy, elevated upper body blood pressures, and diminished blood flow to the lower body. The ductus arteriosus may be open or closed. Coarctation of the aorta is more common in boys and is the leading cause of heart failure in the first few months of life. It’s classified as an obstruction to blood flow leaving the heart.
 |
Echocardiography may show increased left ventricular muscle thickness, coexisting aortic valve abnormalities, and the coarctation site.
Doppler ultrasound and cardiac catheterization evaluate collateral circulation and measure pressure in the right and left ventricles and in the ascending and descending aortas (on both sides of the obstruction).
Aortography locates the site and extent of coarctation.
TREATMENT
For an infant with heart failure caused by coarctation of the aorta, treatment consists of medical management with digoxin, diuretics, oxygen, and sedatives. If medical management fails, surgery may be needed.
The child’s condition usually determines the timing of surgery. Signs of heart failure or hypertension may call for early surgery. If these signs don’t appear, surgery usually occurs during the preschool years.
Before the operation, the child may require endocarditis prophylaxis or, if he’s older and has previously undetected coarctation, antihypertensive therapy. During surgery, the surgeon uses a flap of the left subclavian artery to reconstruct an unobstructed aorta.
Balloon therapy may be indicated for some patients as an alternative to surgical repair. It uses a technique similar to that used to open the coronary arteries, but is performed on the aorta.
SPECIAL CONSIDERATIONS
Palpate the pulses in the legs in newborns and at well-baby visits to detect absent or diminished pulses.
When coarctation in an infant requires rapid digitalization, monitor vital signs closely and watch for digoxin toxicity (poor feeding and vomiting).
Balance intake and output carefully, especially if the infant is receiving diuretics with fluid restriction.
Because the infant may not be able to maintain proper body temperature, regulate environmental temperature with an overbed warmer if needed.
Monitor blood glucose levels to detect possible hypoglycemia, which may occur as glycogen stores become depleted.
Offer the parents emotional support and an explanation of the disorder. Also explain diagnostic procedures, surgery, and drug therapy. Tell parents what to expect postoperatively.
For an older child, assess the blood pressure in his extremities regularly, explain any exercise restrictions, stress the need to take medications properly and to watch for adverse effects, and teach him about tests and other procedures.
After corrective surgery:
Monitor blood pressure closely, using an intra-arterial line. Measure blood pressure in arms and legs. Monitor intake and output.
If the patient develops hypertension and requires nitroprusside or trimethaphan, administer it, as ordered, by continuous I.V. infusion, using an infusion pump. Watch for severe hypotension and regulate the dosage carefully.
Provide pain relief and encourage a gradual increase in activity.
Promote adequate respiratory functioning through turning, coughing, and deep breathing.
Watch for abdominal pain or rigidity and signs of GI or urinary bleeding.
If an older child needs to continue antihypertensives after surgery, teach him and his parents about them.
Stress the importance of continued endocarditis prophylaxis.
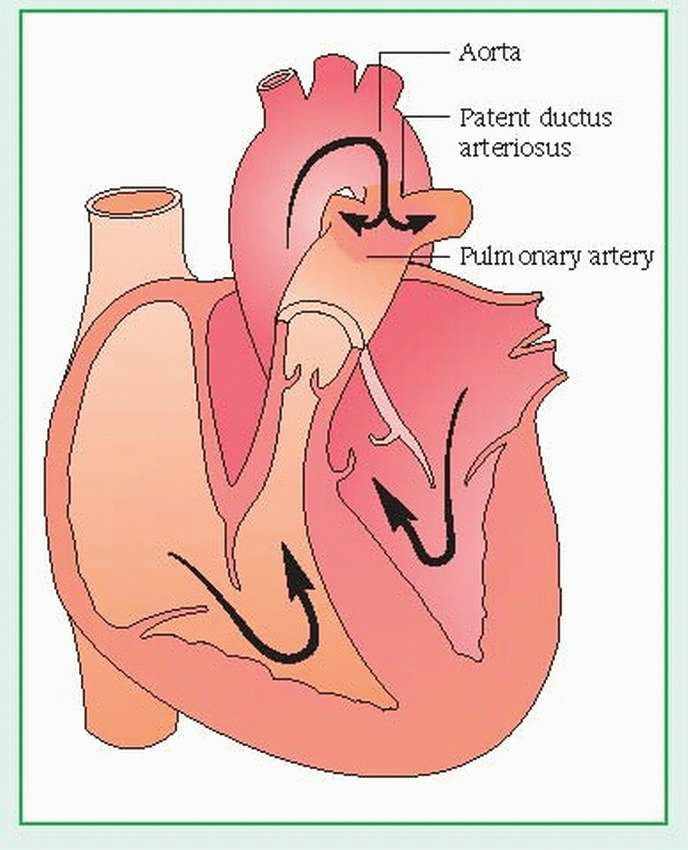
Patent ductus arteriosus
The ductus arteriosus is a fetal blood vessel that connects the pulmonary artery to the descending aorta. In patent ductus arteriosus (PDA), the lumen of the ductus remains open after birth. This creates a left-to-right shunt of blood from the aorta to the pulmonary artery and results in recirculation of arterial blood through the lungs. Initially, PDA may produce no clinical effects, but in time it can precipitate pulmonary vascular disease, causing symptoms to appear by age 40. The prognosis is good if the shunt is small or surgical repair is effective. Otherwise, PDA may advance to intractable heart failure, which may be fatal. (See Understanding patent ductus arteriosus.)
CAUSES AND INCIDENCE
Normally, the ductus closes within days to weeks after birth. Failure to close is most prevalent in premature neonates, probably as a result of abnormalities in oxygenation or the relaxant action of prostaglandin E, which prevents ductal spasm and contracture necessary for closure. PDA commonly accompanies rubella syndrome and may be associated with other congenital defects, such as coarctation of the aorta, ventricular septal defect, and pulmonary and aortic stenoses.
In PDA, relative resistances in pulmonary and systemic vasculature and the size of the ductus determine the amount of left-to-right shunting. The left atrium and left ventricle must accommodate the increased pulmonary venous return, in turn increasing filling pressure and workload on the left side of the heart and possibly causing heart failure. In the final stages of untreated PDA, the left-to-right shunt leads to chronic pulmonary artery hypertension that becomes fixed and unreactive. This causes the shunt to reverse; unoxygenated blood thus enters systemic circulation, causing cyanosis.
PDA is found in 1 of every 2,500 to 5,000 infants and is the most common congenital heart defect found in adults. It affects twice as many females as males.
COMPLICATIONS
Left-sided heart failure
Pulmonary artery hypertension
Respiratory distress (children)
SIGNS AND SYMPTOMS
In neonates, especially those who are premature, a large PDA usually produces respiratory distress, with signs of heart failure due to the tremendous volume of blood shunted to the lungs through a patent ductus and the increased workload on the left side of the heart. Other characteristic features may include heightened susceptibility to respiratory tract infections, slow motor development, and failure to thrive. Most children with PDA have no symptoms except cardiac ones. Others may exhibit signs of heart disease, such as physical underdevelopment, fatigability, and frequent respiratory tract infections. Adults with undetected PDA may develop pulmonary vascular disease and, by age 40, may display fatigability and dyspnea on exertion. About 10% of them also develop infective endocarditis.
Auscultation reveals the classic machinery murmur (Gibson murmur): a continuous murmur (during systole and diastole) best heard at the heart’s base, at the second left intercostal space under the left clavicle in 85% of children with PDA. This murmur may obscure S2. However, with a right-to-left shunt, such a murmur may be absent. Palpation may reveal a thrill at the left sternal border and a prominent left ventricular impulse. Peripheral arterial pulses are bounding (Corrigan’s pulse); pulse pressure is widened because of an elevation in systolic blood pressure and, primarily, a drop in diastolic pressure.
Understanding patent ductus arteriosus
The ductus arteriosus is a fetal blood vessel that connects the pulmonary artery to the descending aorta. Normally, the ductus closes within weeks after birth. However, with patent ductus arteriosus (PDA), it remains open, creating a leftto-right shunt of blood from the aorta to the pulmonary artery and resulting in recirculation of arterial blood through the lungs. PDA is classified as an increased pulmonary blood flow defect.
 |
DIAGNOSIS
Chest X-ray may show increased pulmonary vascular markings, prominent pulmonary arteries, and left ventricle and aorta enlargement.
Electrocardiography (ECG) may be normal or may indicate left atrial or ventricular hypertrophy and, in pulmonary vascular disease, biventricular hypertrophy.
Echocardiography detects and helps estimate the size of a PDA. It also reveals an enlarged left atrium and left ventricle or right ventricular hypertrophy from pulmonary vascular disease.
Cardiac catheterization shows pulmonary arterial oxygen content higher than right ventricular content because of the influx of aortic blood. Increased pulmonary artery pressure indicates a large shunt or, if it exceeds systemic arterial pressure, severe pulmonary vascular disease. Catheterization allows calculation of blood volume crossing the ductus and can rule out associated cardiac defects. Dye injection definitively demonstrates PDA.
TREATMENT
Asymptomatic infants with PDA require no immediate treatment. Those with heart failure require fluid restriction, diuretics, and cardiac glycosides to minimize or control symptoms. If these measures can’t control heart failure, surgery is necessary to ligate the ductus. If symptoms are mild, surgical correction is usually delayed until the infant is between ages 6 months and 3 years, unless problems develop. Before surgery, children with PDA require antibiotics to protect against infective endocarditis. Other forms of therapy include cardiac catheterization to deposit a plug or coil in the ductus to stop shunting or administration of indomethacin I.V. (a prostaglandin inhibitor that’s an alternative to surgery in premature neonates) to induce ductus spasm and closure.
SPECIAL CONSIDERATIONS
PDA necessitates careful monitoring, patient and family teaching, and emotional support.
Watch carefully for signs of PDA in all premature neonates.
Be alert for respiratory distress symptoms resulting from heart failure, which may develop rapidly in a premature neonate. Frequently assess vital signs, ECG, electrolyte levels, and intake and output. Record response to diuretics and other therapy. Watch for signs of digoxin toxicity (poor feeding and vomiting).
If the infant receives indomethacin for ductus closure, watch for possible adverse effects, such as diarrhea, jaundice, bleeding, and renal dysfunction.
Before surgery, carefully explain all treatments and tests to parents. Include the child in your explanations. Arrange for the child and her parents to meet the intensive care unit staff. Tell them about expected I.V. lines, monitoring equipment, and postoperative procedures.
Immediately after surgery, the child may have a central venous pressure catheter and an arterial line in place. Carefully assess vital signs, intake and output, and arterial and venous pressures. Provide pain relief as needed.
Before discharge, review instructions to the parents about activity restrictions based on the child’s tolerance and energy levels. Advise parents not to become overprotective as their child’s tolerance for physical activity increases.
Stress the need for regular follow-up examinations. Advise parents to inform any practitioner who treats their child about his history of surgery for PDA—even if the child is being treated for an unrelated medical problem.
CONGENITAL CYANOTIC DEFECTS
Tetralogy of Fallot
Tetralogy of Fallot is a combination of four cardiac defects: ventricular septal defect (VSD), right ventricular outflow tract obstruction (pulmonary stenosis), right ventricular hypertrophy, and dextroposition of the aorta, with overriding of the VSD. Blood shunts right to left through the VSD, permitting unoxygenated blood to mix with oxygenated blood, resulting in cyanosis. Tetralogy of Fallot sometimes coexists with other congenital heart defects, such as patent ductus arteriosus or atrial septal defect.
CAUSES AND INCIDENCE
The cause of tetralogy of Fallot is unknown, but it results from embryologic hypoplasia of the outflow tract of the right ventricle. Multiple factors, such as Down syndrome, have been associated with its presence. Prenatal risk factors include maternal rubella or other viral illnesses, poor prenatal nutrition, maternal alcoholism, mother older than age 40, and diabetes.
Tetralogy of Fallot occurs in about 5 of every 10,000 infants and accounts for about 10% of all congenital heart diseases. It occurs equally in boys and girls. Before surgical advances made correction possible, about one third of these children died in infancy.
COMPLICATIONS
Cerebral abscess
Pulmonary thrombosis
Venous thrombosis
Cerebral embolism
Infective endocarditis
SIGNS AND SYMPTOMS
The degree of pulmonary stenosis, interacting with the VSD’s size and location, determines the clinical and hemodynamic effects of this complex defect. The VSD usually lies in the right ventricular outflow tract and is generally large enough to permit equalization of right and left ventricular pressures. However, the ratio of systemic vascular resistance to pulmonary stenosis affects the direction and magnitude of shunt flow across the VSD. Severe obstruction of right ventricular outflow produces a right-to-left shunt, causing decreased systemic arterial oxygen saturation, cyanosis, reduced pulmonary blood flow, and hypoplasia of the entire pulmonary vasculature. Increased right ventricular pressure causes right ventricular hypertrophy. Milder forms of pulmonary stenosis result in a left-to-right shunt or no shunt at all.
Generally, the hallmark of the disorder is cyanosis, which usually becomes evident within several months after birth but may be present at birth if the neonate has severe pulmonary stenosis. Between ages 2 months and 2 years, children with tetralogy of Fallot may experience cyanotic or “blue” spells. Such spells result from increased right-to-left shunting, possibly caused by spasm of the right ventricular outflow tract, increased systemic venous return, or decreased systemic arterial resistance.
Exercise, crying, straining, infection, or fever can precipitate blue spells. Blue spells are characterized by dyspnea; deep, sighing respirations; bradycardia; fainting; seizures; and loss of consciousness. Older children may also develop other signs of poor oxygenation, such as clubbing, diminished exercise tolerance, increasing dyspnea on exertion, growth retardation, and eating difficulties. These children habitually squat when they feel short of breath; this is thought to decrease venous return of unoxygenated blood from the legs and increase systemic arterial resistance.
Children with tetralogy of Fallot also risk developing cerebral abscesses, pulmonary thrombosis, venous thrombosis or cerebral embolism, and infective endocarditis.
In females with tetralogy of Fallot who live to childbearing age, the incidence of spontaneous abortion, premature births, and low birth weight rises.
DIAGNOSIS
In a patient with tetralogy of Fallot, auscultation detects a loud systolic heart murmur (best heard along the left sternal border), which may diminish or obscure the pulmonic component of S2. In a patient with a large patent ductus arteriosus, the continuous murmur of the ductus obscures the systolic murmur. Palpation may reveal a cardiac thrill at the left sternal border and an obvious right ventricular impulse. The inferior sternum appears prominent.
The results of special tests also support the diagnosis:
Chest X-ray may demonstrate decreased pulmonary vascular marking, depending on the pulmonary obstruction’s severity, and a bootshaped cardiac silhouette.
Electrocardiography shows right ventricular hypertrophy, right axis deviation and, possibly, right atrial hypertrophy.
Echocardiography identifies septal overriding of the aorta, the VSD, and pulmonary stenosis and detects the hypertrophied walls of the right ventricle.
Laboratory findings reveal diminished arterial oxygen saturation and polycythemia (hematocrit may be more than 60%) if the cyanosis is severe and long-standing, predisposing the patient to thrombosis.
Cardiac catheterization confirms the diagnosis by visualizing pulmonary stenosis, the VSD, and the overriding aorta and ruling out other cyanotic heart defects. This test also measures the degree of oxygen saturation in aortic blood
TREATMENT
Effective management of tetralogy of Fallot necessitates prevention and treatment of complications, measures to relieve cyanosis, and palliative or corrective surgery. During cyanotic spells, the knee-chest position and administration of oxygen and morphine improve oxygenation. Propranolol (a beta-adrenergic blocking agent) may prevent blue spells.
Palliative surgery is performed on infants with potentially fatal hypoxic spells. The goal of surgery is to enhance blood flow to the lungs to reduce hypoxia; this is often accomplished by joining the subclavian artery to the pulmonary artery (Blalock-Taussig procedure). Supportive measures include prophylactic antibiotics to prevent infective endocarditis or cerebral abscess administered before, during, and after bowel, bladder, or any other surgery or dental treatments. Management may also include phlebotomy in children with polycythemia.
Complete corrective surgery to relieve pulmonary stenosis and close the VSD, directing left ventricular outflow to the aorta, requires cardiopulmonary bypass with hypothermia to decrease oxygen utilization during surgery, especially in young children. An infant may have this corrective surgery without prior palliative surgery. It’s usually done when progressive hypoxia and polycythemia impair the quality of his life, rather than at a specific age. However, most children require surgery before they reach school age.
SPECIAL CONSIDERATIONS
Explain tetralogy of Fallot to the parents. Inform them that their child will set his own exercise limits and will know when to rest. Make sure they understand that their child can engage in physical activity, and advise them not to be overprotective.
Teach the parents to recognize serious hypoxic spells, which can dramatically increase cyanosis; deep, sighing respirations; and loss of consciousness. Tell them to place their child in the kneechest position and to report such spells immediately. Emergency treatment may be necessary.
Instruct the parents on ways to prevent overexerting their child, such as feeding him slowly and providing smaller and more frequent meals. Tell them that remaining calm may decrease his anxiety and that anticipating his needs may minimize crying. Encourage the parents to recruit other family members in the care of the child to help prevent their own exhaustion.
To prevent infective endocarditis and other infections, warn the parents to keep their child away from people with infections. Urge them to encourage good dental hygiene, and tell them to watch for ear, nose, and throat infections and dental caries, all of which necessitate immediate treatment. When dental care, infections, or surgery requires prophylactic antibiotics, tell the parents to make sure the child completes the prescribed regimen.
If the child requires medical attention for an unrelated problem, advise the parents to inform the practitioner immediately of the child’s history of tetralogy of Fallot because any treatment must take this serious heart defect into consideration.
During hospitalization, alert the staff to the child’s condition. Because of the right-to-left shunt through the VSD, treat I.V. lines like arterial lines. A clot dislodged from a catheter tip in a vein can cross the VSD and cause cerebral embolism. The same thing can happen if air enters the venous lines.
After palliative surgery:
Monitor oxygenation and arterial blood gas (ABG) values closely in the intensive care unit.
If the child has undergone the Blalock-Taussig procedure, don’t use the arm on the operative side for measuring blood pressure, inserting I.V. lines, or drawing blood samples, because blood perfusion on this side diminishes greatly until collateral circulation develops. Note this on the child’s chart and at his bedside.
After corrective surgery:
Watch for right bundle-branch block or more serious disturbances of atrioventricular conduction and for ventricular ectopic beats.
Be alert for other postoperative complications, such as bleeding, right-sided heart failure, and respiratory failure. After surgery, transient heart failure is common and may require treatment with digoxin and diuretics.
Monitor left atrial pressure directly. A pulmonary artery catheter may also be used to check central venous and pulmonary artery pressures.
Frequently check color and vital signs. Obtain ABG measurements regularly to assess oxygenation. Suction to prevent atelectasis and pneumonia, as needed. Monitor mechanical ventilation.
Monitor and record intake and output accurately.
If atrioventricular block develops with a low heart rate, a temporary external pacemaker may be necessary.
If blood pressure or cardiac output is inadequate, catecholamines may be ordered by continuous I.V. infusion. To decrease left ventricular workload, administer nitroprusside, if ordered, and provide analgesics, as needed.
Keep the parents informed about their child’s progress. After discharge, the child may require digoxin, diuretics, and other drugs. Stress the importance of complying with the prescribed regimen and make sure the parents know how and when to administer these medications. Teach the parents to watch for signs of digoxin toxicity (anorexia, nausea, and vomiting). Prophylactic antibiotics to prevent infective endocarditis will still be required. Advise the parents to avoid becoming overprotective as the child’s tolerance for physical activity rises.
Transposition of the great arteries
In this congenital heart defect, the great arteries are reversed: the aorta arises from the right ventricle and the pulmonary artery from the left ventricle, producing two noncommunicating circulatory systems (pulmonary and systemic). Transposition accounts for about 5% of all congenital heart defects and often coexists with other congenital heart defects, such as ventricular septal defect (VSD), VSD with pulmonary stenosis (PS), atrial septal defect (ASD), and patent ductus arteriosus (PDA). It affects two to three times more males than females.
CAUSES AND INCIDENCE
Transposition of the great arteries results from faulty embryonic development, but the cause of such development is unknown. In transposition, oxygenated blood returning to the left side of the heart is carried back to the lungs by a transposed pulmonary artery; unoxygenated blood returning to the right side of the heart is carried to the systemic circulation by a transposed aorta.
Communication between the pulmonary and systemic circulations is necessary for survival. In infants with isolated transposition, blood mixes only at the patent foramen ovale and at the PDA, resulting in slight mixing of unoxygenated systemic blood and oxygenated pulmonary blood. In infants with concurrent cardiac defects, greater mixing of blood occurs.
Transposition of the great arteries occurs in about 40 of every 100,000 infants.
COMPLICATIONS
Chronic heart failure
Poor oxygenation
Arrhythmias
Right-sided heart failure
SIGNS AND SYMPTOMS
Within the first few hours after birth, neonates with transposition of the great arteries and no other heart defects generally show cyanosis and tachypnea, which worsen with crying. After several days or weeks, such neonates usually develop signs of heart failure (gallop rhythm, tachycardia, dyspnea, hepatomegaly, and cardiomegaly). S2 is louder than normal because the anteriorly transposed aorta is directly behind the sternum; in many cases, however, no murmur can be heard during the first few days of life. Associated defects (ASD, VSD, or PDA) cause their
typical murmurs and may minimize cyanosis but may also cause other complications (especially severe heart failure). VSD with PS produces a characteristic murmur and severe cyanosis.
typical murmurs and may minimize cyanosis but may also cause other complications (especially severe heart failure). VSD with PS produces a characteristic murmur and severe cyanosis.
As infants with this defect grow older, cyanosis is their most prominent abnormality. However, they also develop diminished exercise tolerance, fatigability, coughing, clubbing, and more pronounced murmurs if ASD, VSD, PDA, or PS is present.
DIAGNOSIS
Chest X-rays are normal in the first days of life. Within days to weeks, right atrial and right ventricular enlargement characteristically cause the heart to appear oblong. X-rays also show increased pulmonary vascular markings, except when PS coexists.
Electrocardiography typically reveals right axis deviation and right ventricular hypertrophy but may be normal in a neonate.
Echocardiography demonstrates the reversed position of the aorta and pulmonary artery and records echoes from both semilunar valves simultaneously, due to aortic valve displacement. It also detects other cardiac defects. Cardiac catheterization reveals decreased oxygen saturation in left ventricular blood and aortic blood; increased right atrial, right ventricular, and pulmonary artery oxygen saturation; and right ventricular systolic pressure equal to systemic pressure. Dye injection reveals the transposed vessels and the presence of any other cardiac defects
♦ Arterial blood gas (ABG) measurements indicate hypoxia and secondary metabolic acidosis.
TREATMENT
An infant with transposition may undergo atrial balloon septostomy (Rashkind procedure) during cardiac catheterization. This procedure enlarges the patent foramen ovale, which improves oxygenation by allowing greater mixing of the pulmonary and systemic circulations. Atrial balloon septostomy requires passage of a balloon-tipped catheter through the foramen ovale and subsequent inflation and withdrawal across the atrial septum. This procedure alleviates hypoxia to a certain degree. Afterward, digoxin and diuretics can lessen heart failure until the infant is ready to withstand corrective surgery (usually between birth and age 1).
One of three surgical procedures can correct transposition, depending on the defect’s physiology. The Mustard procedure replaces the atrial septum with a Dacron or pericardial partition that allows systemic venous blood to be channeled to the pulmonary artery—which carries the blood to the lungs for oxygenation— and oxygenated blood returning to the heart to be channeled from the pulmonary veins into the aorta. The Senning procedure accomplishes the same result, using the atrial septum to create partitions to redirect blood flow. In the arterial switch, or Jantene procedure, transposed arteries are surgically anastomosed to the correct ventricle. For this procedure to be successful, the left ventricle must be used to pump at systemic pressure, as it does in neonates or in children with a left ventricular outflow obstruction or a large VSD. The Jantene procedure is the procedure of choice; however, the Mustard and Senning procedures may be used when specific anatomic conditions exist.
SPECIAL CONSIDERATIONS
Explain cardiac catheterization and all necessary procedures to the parents. Offer emotional support.
Monitor vital signs, ABG values, urine output, and central venous pressure, watching for signs of heart failure. Give digoxin and I.V. fluids, being careful to avoid fluid overload.
Teach the parents to recognize signs of heart failure and digoxin toxicity (poor feeding and vomiting). Stress the importance of regular checkups to monitor cardiovascular status.
Teach the parents to protect their infant from infection and to give antibiotics.
Tell the parents to let their child develop normally. They need not restrict activities; he’ll set his own limits.
If the patient is scheduled for surgery, explain the procedure to the parents and child, if old enough. Teach them about the intensive care unit and introduce them to the staff. Also explain postoperative care.
Preoperatively, monitor ABG values, acidbase balance, intake and output, and vital signs.
After corrective surgery:
Monitor cardiac output by checking blood pressure, skin color, heart rate, urine output, central venous and left atrial pressures, and level of consciousness. Report abnormalities or changes.
Carefully monitor ABG levels and report changes in trends.
To detect supraventricular conduction blocks and arrhythmias, monitor the patient closely. Watch for signs of atrioventricular blocks, atrial arrhythmias, and faulty sinoatrial function.
After the Mustard or Senning procedure, watch for signs of baffle obstruction such as marked facial edema.
Encourage parents to help their child assume new activity levels and independence. Teach them about postoperative antibiotic prophylaxis for endocarditis.
ACQUIRED INFLAMMATORY HEART DISEASE
Myocarditis
Myocarditis is focal or diffuse inflammation of the cardiac muscle (myocardium). It may be acute or chronic and can occur at any age. In many cases, myocarditis fails to produce specific cardiovascular symptoms or electrocardiogram (ECG) abnormalities, and recovery is usually spontaneous, without residual defects. Occasionally, myocarditis is complicated by heart failure; in rare cases, it leads to cardiomyopathy.
CAUSES AND INCIDENCE
Myocarditis may result from:
bacterial infections—diphtheria; tuberculosis; typhoid fever; tetanus; and staphylococcal, pneumococcal, and gonococcal infections
chemical poisons—such as chronic alcoholism
helminthic infections—such as trichinosis
hypersensitive immune reactions—acute rheumatic fever and postcardiotomy syndrome
parasitic infections—especially South American trypanosomiasis (Chagas’ disease) in infants and immunosuppressed adults; also toxoplasmosis
radiation therapy—large doses of radiation to the chest in treating lung or breast cancer
viral infections (most common cause in the United States and western Europe)—coxsackievirus A and B strains and, possibly, poliomyelitis, influenza, rubeola, rubella, and adenoviruses and echoviruses
Myocarditis occurs in 1 to 10 of every 100,000 people in the United States. The median age for this disorder is 42, and incidence is equal between males and females. Children, especially neonates, and persons who are immunocompromised or pregnant (especially pregnant black women) are at higher risk for developing this disorder.
COMPLICATIONS
Arrhythmias
Thromboembolism
Chronic valvulitis (when disease results from rheumatic fever)
Recurrence of disease
Left-sided heart failure (occasional) Cardiomyopathy (rare)
SIGNS AND SYMPTOMS
Myocarditis usually causes nonspecific symptoms—such as fatigue, dyspnea, palpitations, and fever—that reflect the accompanying systemic infection. Occasionally, it may produce mild, continuous pressure or soreness in the chest (unlike the recurring, stress-related pain of angina pectoris). Although myocarditis is usually self-limiting, it may induce myofibril degeneration that results in right- and left-sided heart failure, with cardiomegaly, jugular vein distention, dyspnea, persistent fever with resting or exertional tachycardia disproportionate to the degree of fever, and supraventricular and ventricular arrhythmias. Sometimes myocarditis recurs or produces chronic valvulitis (when it results from rheumatic fever), cardiomyopathy, arrhythmias, and thromboembolism.
DIAGNOSIS
Patient history commonly reveals recent febrile upper respiratory tract infection, viral pharyngitis, or tonsillitis. Physical examination shows supraventricular and ventricular arrhythmias, S3 and S4 gallops, a faint S1, possibly a murmur of mitral insufficiency (from papillary muscle dysfunction) and, if pericarditis is present, a pericardial friction rub.
Laboratory tests can’t unequivocally confirm myocarditis, but the following findings support this diagnosis:
cardiac enzymes: elevated creatine kinase (CK), CK-MB, aspartate aminotransferase, and lactate dehydrogenase levels
increased white blood cell count and erythrocyte sedimentation rate
elevated antibody titers (such as antistreptolysin-O titer in rheumatic fever)
Endomyocardial biopsy is rarely performed to diagnose myocarditis; the procedure is invasive and costly. A negative biopsy doesn’t exclude the diagnosis, and a repeat biopsy may be needed.
ECG typically shows diffuse ST-segment and T-wave abnormalities as in pericarditis, conduction defects (prolonged PR interval), and other supraventricular arrhythmias. Echocardiography demonstrates some degree of left ventricular
dysfunction, and radionuclide scanning may identify inflammatory and necrotic changes characteristic of myocarditis.
dysfunction, and radionuclide scanning may identify inflammatory and necrotic changes characteristic of myocarditis.
Stool and throat cultures may identify bacteria.
TREATMENT
Treatment includes antibiotics for bacterial infection, modified bed rest to decrease cardiac workload, and careful management of complications. Inotropic support of cardiac function with amrinone, dopamine, or dobutamine may be needed. Heart failure requires restriction of activity to minimize myocardial oxygen consumption, supplemental oxygen therapy, sodium restriction, diuretics to decrease fluid retention, and cardiac glycosides to increase myocardial contractility. However, cardiac glycosides should be administered cautiously because some patients with myocarditis may show a paradoxical sensitivity to even small doses. Arrhythmias necessitate prompt but cautious administration of antiarrhythmics because these drugs depress myocardial contractility. Thromboembolism requires anticoagulation therapy. Treatment with corticosteroids or other immunosuppressants may be used to reduce inflammation, but they haven’t been shown to change the progression of myocarditis. Nonsteroidal anti-inflammatory drugs are contraindicated during the acute phase (first 2 weeks) because they increase myocardial damage.
Surgical treatment may include left ventricular assistive devices and extracorporeal membrane oxygenation for support of cardiogenic shock. Cardiac transplantation has been beneficial for giant cell myocarditis.
SPECIAL CONSIDERATIONS
Assess cardiovascular status frequently, watching for signs of heart failure, such as dyspnea, hypotension, and tachycardia. Check for changes in cardiac rhythm or conduction.
Observe for signs of digoxin toxicity (anorexia, nausea, vomiting, blurred vision, and cardiac arrhythmias) and for complicating factors that may potentiate toxicity, such as electrolyte imbalance or hypoxia.
Stress the importance of bed rest. Assist with bathing, as necessary; provide a bedside commode because this stresses the heart less than using a bedpan. Reassure the patient that activity limitations are temporary. Offer diversional activities that are physically undemanding.
During recovery, recommend that the patient resume normal activities slowly and avoid competitive sports.
Instruct patient to obtain prompt treatment of causative disorders
Instruct patient to practice good hygiene, including thorough hand-washing.
Tell patient to thoroughly wash and cook food.
Endocarditis
Endocarditis (also known as infective or bacterial endocarditis) is an infection of the endocardium, heart valves, or cardiac prostheses resulting from bacterial or fungal invasion. This invasion produces vegetative growths on the heart valves, endocardial lining of a heart chamber, or endothelium of a blood vessel that may embolize to the spleen, kidneys, central nervous system, and lungs. In endocarditis, fibrin and platelets aggregate on the valve tissue and engulf circulating bacteria or fungi that flourish and produce friable verrucous vegetations. (See Degenerative changes in endocarditis, page 22.) Such vegetations may cover the valve surfaces, causing ulceration and necrosis; they may also extend to the chordae tendineae, leading to their rupture and subsequent valvular insufficiency. Untreated endocarditis is usually fatal, but with proper treatment, 70% of patients recover. The prognosis is worst when endocarditis causes severe valvular damage, leading to insufficiency and heart failure, or when it involves a prosthetic valve.
CAUSES AND INCIDENCE
Most cases of endocarditis occur in I.V. drug abusers, patients with prosthetic heart valves, and those with mitral valve prolapse (especially males with a systolic murmur). These conditions have surpassed rheumatic heart disease as the leading risk factor. Other predisposing conditions include coarctation of the aorta, tetralogy of Fallot, subaortic and valvular aortic stenosis, ventricular septal defects, pulmonary stenosis, Marfan syndrome, degenerative heart disease (especially calcific aortic stenosis) and, rarely, syphilitic aortic valve. However, some patients with endocarditis have no underlying heart disease.
Infecting organisms differ among these groups. In patients with native valve endocarditis who aren’t I.V. drug abusers, causative organisms usually include—in order of frequency—streptococci (especially Streptococcus viridans), staphylococci, or enterococci.
Although many other bacteria occasionally cause the disorder, fungal causes are rare in this group. The mitral valve is involved most commonly, followed by the aortic valve.
In patients who are I.V. drug abusers, Staphylococcus aureus is the most common infecting organism. Less commonly, streptococci, enterococci, gram-negative bacilli, or fungi cause the disorder. The tricuspid valve is involved most commonly, followed by the aortic and then the mitral valve.
In patients with prosthetic valve endocarditis, early cases (those that develop within 60 days of valve insertion) are usually due to staphylococcal infection. However, gram-negative aerobic organisms, fungi, streptococci, enterococci, or diphtheroids may also cause the disorder. The course is usually fulminant and is associated with a high mortality. Late cases (occurring after 60 days) present similarly to native valve endocarditis.
In the United States, endocarditis affects 1.4 to 4.2 people out of every 100,000. Males are twice as likely as females to acquire this infection, and the mean age of onset is 50. Mortality is associated with increased age, infection of the aortic valve, heart failure and underlying heart disease, and central nervous system complications; mortality rates vary with the infecting organism.
COMPLICATIONS
Left-sided heart failure
Valvular stenosis or insufficiency
Myocardial erosion
SIGNS AND SYMPTOMS
Early clinical features of endocarditis are usually nonspecific and include malaise, weakness, fatigue, weight loss, anorexia, arthralgia, night sweats, chills, valvular insufficiency and, in 90% of patients, an intermittent fever that may recur for weeks. A more acute onset is associated with organisms of high pathogenicity such as S. aureus. Endocarditis commonly causes a loud, regurgitant murmur typical of the underlying heart lesion. A suddenly changing murmur or the discovery of a new murmur in the presence of fever is a classic physical sign of endocarditis.
In about 30% of patients, embolization from vegetating lesions or diseased valvular tissue may produce typical features of splenic, renal, cerebral, or pulmonary infarction or of peripheral vascular occlusion:
splenic infarction—pain in the left upper quadrant, radiating to the left shoulder, and abdominal rigidity
renal infarction—hematuria, pyuria, flank pain, and decreased urine output
cerebral infarction—hemiparesis, aphasia, or other neurologic deficits
pulmonary infarction (most common in rightsided endocarditis, which commonly occurs among I.V. drug abusers and after cardiac surgery)—cough, pleuritic pain, pleural friction rub, dyspnea, and hemoptysis
peripheral vascular occlusion—numbness and tingling in an arm, leg, finger, or toe, or signs of impending peripheral gangrene
Other signs may include splenomegaly; petechiae of the skin (especially common on the upper anterior trunk) and the buccal, pharyngeal, or conjunctival mucosa; and splinter hemorrhages under the nails. Rarely, endocarditis produces Osler’s nodes (tender, raised, subcutaneous lesions on the fingers or toes), Roth’s spots (hemorrhagic areas with white centers on the retina), and Janeway lesions (purplish macules on the palms or soles).
DIAGNOSIS
Any patient who is at risk for or susceptible to endocarditis, such as those with artificial heart valves or other predisposing factors, should have prophylactic antibiotics before dental or other invasive procedures.
In addition, the patient should practice good hygiene, including thoroughly washing his hands and washing fruits and vegetables and thoroughly cooking all food to prevent introducing organisms into his system. Maintaining good oral health by daily brushing and flossing and having regular dental checkups can also prevent infection. Be sure to advise the patient to notify his family practitioner as well as his dentist or another specialist that he has a condition that places him at high risk for endocarditis.
The remaining 10% may have negative blood cultures, possibly suggesting fungal infection or infections that are difficult to diagnose, such as Haemophilus parainfluenzae.
Other abnormal but nonspecific laboratory test results include:
normal or elevated white blood cell count
abnormal histiocytes (macrophages)
elevated erythrocyte sedimentation rate
normocytic, normochromic anemia (in 70% to 90% of patients)
proteinuria and microscopic hematuria (in about 50% of patients)
positive serum rheumatoid factor (in about 50% of patients after endocarditis is present for 3 to 6 weeks)
Echocardiography (particularly, transesophageal) may identify valvular damage; electrocardiography may show atrial fibrillation and other arrhythmias that accompany valvular disease.
TREATMENT
The goal of treatment is to eradicate the infecting organism with appropriate antimicrobial therapy, which should start promptly and continue over 4 to 6 weeks. Selection of an antibiotic is based on identification of the infecting organism and on sensitivity studies. While awaiting results, or if blood cultures are negative, empiric antimicrobial therapy is based on the likely infecting organism.
Supportive treatment includes bed rest, aspirin for fever and aches, and sufficient fluid intake. Severe valvular damage, especially aortic or mitral insufficiency, may require corrective surgery if refractory heart failure develops, or in cases requiring that an infected prosthetic valve be replaced.
SPECIAL CONSIDERATIONS
Before giving antibiotics, obtain a patient history of allergies. Administer antibiotics on time to maintain consistent antibiotic blood levels.
Observe for signs of infiltration or inflammation at the venipuncture site, possible complications of long-term I.V. drug administration. To reduce the risk of these complications, rotate venous access sites.
Watch for signs of embolization (hematuria, pleuritic chest pain, left upper quadrant pain, or paresis), a common occurrence during the first 3 months of treatment. Tell the patient to watch for and report these signs, which may indicate impending peripheral vascular occlusion or splenic, renal, cerebral, or pulmonary infarction.
Monitor the patient’s renal status (blood urea nitrogen levels, creatinine clearance, and urine output) to check for signs of renal emboli or evidence of drug toxicity.
Observe for signs of heart failure, such as dyspnea, tachypnea, tachycardia, crackles, jugular vein distention, edema, and weight gain.
Provide reassurance by teaching the patient and his family about this disease and the need for prolonged treatment. Tell them to watch closely for fever, anorexia, and other signs of relapse about 2 weeks after treatment stops. Suggest quiet diversionary activities to prevent excessive physical exertion.
Make sure susceptible patients understand the need for prophylactic antibiotics before, during, and after dental work, childbirth, and genitourinary, GI, or gynecologic procedures.
Teach patients how to recognize symptoms of endocarditis and tell them to notify the practitioner at once if such symptoms occur. (See Preventing endocarditis.)
Patterns of cardiac pain
Although pain perception is individualistic, specific characteristics are associated with different types of cardiac pain, as shown below.
Pericarditis
Onset and duration
▪ Sudden onset; continuous pain lasting for days; residual soreness
Location and radiation
▪ Substernal pain to left of midline; radiation to back or subclavicular area
Quality and intensity
▪ Mild ache to severe pain, deep or superficial; “stabbing,” “knifelike”
Signs and symptoms
▪ Precordial friction rub; increased pain with movement, inspiration, laughing, coughing; decreased pain with sitting or leaning forward (sitting up pulls heart away from diaphragm)
Precipitating factors
▪ Myocardial infarction or upper respiratory tract infection; invasive cardiac trauma
Angina
Onset and duration
▪ Gradual or sudden onset; pain usually lasts less than 15 minutes and not more than 30 minutes (average: 3 minutes)
Location and radiation
▪ Substernal or anterior chest pain, not sharply localized; radiation to back, neck, arms, jaws, even upper abdomen or fingers
Quality and intensity
▪ Mild-to-moderate pressure; deep sensation; varied pattern of attacks; “tightness,” “squeezing,” “crushing,” “pressure”
Signs and symptoms
▪ Dyspnea, diaphoresis, nausea, desire to void, belching, apprehension
Precipitating factors
▪ Exertion, stress, eating, cold or hot and humid weather
Myocardial infarction
Onset and duration
▪ Sudden onset; pain lasts 30 minutes to 2 hours; waxes and wanes; residual soreness 1 to 3 days
Location and radiation
▪ Substernal, midline, or anterior chest pain; radiation to jaws, neck, back, shoulders, or one or both arms
Quality and intensity
▪ Persistent, severe pressure; deep sensation; “crushing,” “squeezing,” “heavy,” “oppressive”
Signs and symptoms
▪ Nausea, vomiting, apprehension, dyspnea, diaphoresis, increased or decreased blood pressure; gallop heart sound, “sensation of impending doom”
Precipitating factors
▪ Occurrence at rest or during physical exertion or emotional stress
Pericarditis
Pericarditis is an inflammation of the pericardium, the fibroserous sac that envelops, supports, and protects the heart. It occurs in both acute and chronic forms. Acute pericarditis can be fibrinous or effusive, with purulent serous or hemorrhagic exudate; chronic constrictive pericarditis is characterized by dense fibrous pericardial thickening. The prognosis depends on the underlying cause but is generally good in acute pericarditis, unless constriction occurs.
CAUSES AND INCIDENCE
Common causes of this disease include:
bacterial, fungal, or viral infection (infectious pericarditis)
neoplasms (primary or metastatic from lungs, breasts, or other organs)
high-dose radiation to the chest
uremia
hypersensitivity or autoimmune disease, such as acute rheumatic fever (most common cause of pericarditis in children), systemic lupus erythematosus, and rheumatoid arthritis
postcardiac injury such as myocardial infarction (MI), which later causes an autoimmune reaction (Dressler’s syndrome) in the pericardium; trauma; or surgery that leaves the pericardium intact but causes blood to leak into the pericardial cavity
drugs, such as hydralazine or procainamide
idiopathic factors (most common in acute pericarditis)
Less common causes include aortic aneurysm with pericardial leakage and myxedema with cholesterol deposits in the pericardium.
COMPLICATIONS
Pericardial effusion
Cardiac tamponade
Shock
Cardiovascular collapse
Death
SIGNS AND SYMPTOMS
Acute pericarditis typically produces a sharp and often sudden pain that usually starts over the sternum and radiates to the neck, shoulders, back, and arms. However, unlike the pain of MI, pericardial pain is often pleuritic, increasing with deep inspiration and decreasing when the patient sits up and leans forward, pulling the heart away from the diaphragmatic pleurae of the lungs.
Pericardial effusion, the major complication of acute pericarditis, may produce effects of heart failure (such as dyspnea, orthopnea, and tachycardia), ill-defined substernal chest pain, and a feeling of fullness in the chest. (See Patterns of cardiac pain.)
If the fluid accumulates rapidly, cardiac tamponade may occur, resulting in pallor, clammy skin, hypotension, pulsus paradoxus (a decrease in systolic blood pressure of 15 mm Hg or more during slow inspiration), jugular vein distention and, eventually, cardiovascular collapse and death.
Chronic constrictive pericarditis causes a gradual increase in systemic venous pressure and produces symptoms similar to those of chronic right-sided heart failure (fluid retention, ascites, and hepatomegaly).
DIAGNOSIS
Because pericarditis commonly coexists with other conditions, diagnosis of acute pericarditis depends on typical clinical features and elimination of other possible causes. The pericardial friction rub, a classic symptom, is a grating sound heard as the heart moves. It can usually be auscultated best during forced expiration, while the patient leans forward or is on his hands and knees in bed. It may have up to three components, corresponding to the timing of atrial systole, ventricular systole, and the rapid-filling phase of ventricular diastole. Occasionally, this friction rub is heard only briefly or not at all. Nevertheless, its presence, together with other characteristic features, is diagnostic of acute pericarditis. In addition, if acute pericarditis has caused very large pericardial effusions, physical examination reveals increased cardiac dullness and diminished or absent apical impulse and distant heart sounds.
Chest X-ray, echocardiogram, chest magnetic resonance imaging (MRI), heart MRI, heart computed tomography scan, and radionuclide scanning can detect fluid that has accumulated in the pericardial sac. They may also show enlargement of the heart and signs of inflammation or scarring, depending on the cause of pericarditis.
In patients with chronic pericarditis, acute inflammation or effusions don’t occur—only restricted cardiac filling.
Laboratory results reflect inflammation and may identify its cause:
normal or elevated white blood cell count, especially in infectious pericarditis
elevated erythrocyte sedimentation rate
slightly elevated cardiac enzyme levels with associated myocarditis
culture of pericardial fluid obtained by open surgical drainage or cardiocentesis (sometimes identifies a causative organism in bacterial or fungal pericarditis)
electrocardiography showing the following changes in acute pericarditis: elevation of ST segments in the standard limb leads and most precordial leads without the significant changes in QRS morphology that occur with MI, atrial ectopic rhythms such as atrial fibrillation and, in pericardial effusion, diminished QRS voltage
Other pertinent laboratory data include blood urea nitrogen levels to check for uremia, antistreptolysin-O titers to detect rheumatic fever, and a purified protein derivative skin test to check for tuberculosis. In pericardial effusion, echocardiography is diagnostic when it shows an echo-free space between the ventricular wall and the pericardium.
TREATMENT
The goal of treatment is to relieve symptoms and manage the underlying systemic disease. In acute idiopathic pericarditis and postthoracotomy pericarditis, treatment consists of bed rest as long as fever and pain persist, and nonsteroidal drugs, such as aspirin and indomethacin, to relieve pain and reduce inflammation. Post-MI patients should avoid nonsteroidal anti-inflammatory drugs and steroids because they may interfere with myocardial scar formation. If these drugs fail to relieve symptoms, corticosteroids may be used. Although corticosteroids produce rapid and effective relief, they must be used cautiously because episodes may recur when therapy is discontinued.
Infectious pericarditis that results from disease of the left pleural space, mediastinal abscesses, or septicemia requires antibiotics (possibly by direct pericardial injection), surgical drainage, or both. Cardiac tamponade may
require pericardiocentesis. Signs of tamponade include pulsus paradoxus, jugular vein distention, dyspnea, and shock.
require pericardiocentesis. Signs of tamponade include pulsus paradoxus, jugular vein distention, dyspnea, and shock.
Recurrent pericarditis may necessitate partial pericardectomy, which creates a “window” that allows fluid to drain into the pleural space. In constrictive pericarditis, total pericardectomy to permit adequate filling and contraction of the heart may be necessary. Treatment must also include management of rheumatic fever, uremia, tuberculosis, and other underlying disorders.
SPECIAL CONSIDERATIONS
A patient with pericarditis needs complete bed rest. In addition, health care includes:
assessing pain in relation to respiration and body position to distinguish pericardial pain from myocardial ischemic pain
placing the patient in an upright position to relieve dyspnea and chest pain; providing analgesics and oxygen; and reassuring the patient with acute pericarditis that his condition is temporary and treatable
monitoring for signs of cardiac compression or cardiac tamponade, possible complications of pericardial effusion (Signs include decreased blood pressure, increased central venous pressure, and pulsus paradoxus. Because cardiac tamponade requires immediate treatment, keep a pericardiocentesis set handy whenever pericardial effusion is suspected.)
explaining tests and treatments to the patient (If surgery is necessary, he should learn deepbreathing and coughing exercises beforehand. Postoperative care is similar to that given after cardiothoracic surgery.)
Rheumatic fever and rheumatic heart disease
Acute rheumatic fever is a systemic inflammatory disease of childhood, in many cases recurrent, that follows a group A beta-hemolytic streptococcal infection. Rheumatic heart disease refers to the cardiac manifestations of rheumatic fever and includes pancarditis (myocarditis, pericarditis, and endocarditis) during the early acute phase and chronic valvular disease later. Long-term antibiotic therapy can minimize the recurrence of rheumatic fever, reducing the risk of permanent cardiac damage and eventual valvular deformity. However, severe pancarditis occasionally produces fatal heart failure during the acute phase. Of the patients who survive this complication, about 20% die within 10 years.
CAUSES AND INCIDENCE
Rheumatic fever appears to be a hypersensitivity reaction to a group A beta-hemofytic streptococcal infection, in which antibodies manufactured to combat streptococci react and produce characteristic lesions at specific tissue sites, especially in the heart and joints. Because very few persons (3%) with streptococcal infections ever contract rheumatic fever, altered host resistance must be involved in its development or recurrence. Although rheumatic fever tends to be familial, this may merely reflect contributing environmental factors. For example, in lower socioeconomic groups, incidence is highest in children between ages 5 and 15, probably as a result of malnutrition and crowded living conditions. This disease strikes generally during cool, damp weather in the winter and early spring. In the United States, it’s most common in the northern states.
COMPLICATIONS
Destruction of mitral and aortic valves
Severe pancarditis
Pericardial effusion
Fatal heart failure
SIGNS AND SYMPTOMS
In 95% of patients, rheumatic fever characteristically follows a streptococcal infection that appeared a few days to 6 weeks earlier. A temperature of at least 100.4° F (38° C) occurs, and most patients complain of migratory joint pain or polyarthritis. Swelling, redness, and signs of effusion usually accompany such pain, which most commonly affects the knees, ankles, elbows, or hips. In 5% of patients (generally those with carditis), rheumatic fever causes skin lesions such as erythema marginatum, a nonpruritic, macular, transient rash that gives rise to red lesions with blanched centers. Rheumatic fever may also produce firm, movable, nontender, subcutaneous nodules about 3 mm to 2 cm in diameter, usually near tendons or bony prominences of joints (especially the elbows, knuckles, wrists, and knees) and less often on the scalp and backs of the hands. These nodules persist for a few days to several weeks and, like erythema marginatum, often accompany carditis.
Later, rheumatic fever may cause transient chorea, which develops up to 6 months after the original streptococcal infection. Mild chorea may produce hyperirritability, a deterioration in handwriting, or an inability to concentrate. Severe chorea (Sydenham’s chorea) causes purposeless, nonrepetitive, involuntary muscle spasms; poor muscle coordination; and
weakness. Chorea always resolves without residual neurologic damage.
weakness. Chorea always resolves without residual neurologic damage.
The most destructive effect of rheumatic fever is carditis, which develops in up to 50% of patients and may affect the endocardium, myocardium, pericardium, or the heart valves. Pericarditis causes a pericardial friction rub and, occasionally, pain and effusion. Myocarditis produces characteristic lesions called Aschoff bodies (in the acute stages) and cellular swelling and fragmentation of interstitial collagen, leading to formation of a progressively flbrotic nodule and interstitial scars. Endocarditis causes valve leaflet swelling, erosion along the lines of leaflet closure, and blood, platelet, and fibrin deposits, which form beadlike vegetations. Endocarditis affects the mitral valve most often in females and the aortic valve most often in males. In both females and males, endocarditis affects the tricuspid valves occasionally and the pulmonic valve only rarely.
Severe rheumatic carditis may cause heart failure with dyspnea; right upper quadrant pain; tachycardia; tachypnea; a hacking, nonproductive cough; edema; and significant mitral and aortic murmurs. The most common of such murmurs include:
a systolic murmur of mitral insufficiency (highpitched, blowing, holosystolic, loudest at apex, possibly radiating to the anterior axillary line)
a midsystolic murmur due to stiffening and swelling of the mitral leaflet
occasionally, a diastolic murmur of aortic insufficiency (low-pitched, rumbling, almost inaudible). Valvular disease may eventually result in chronic valvular stenosis and insufficiency, including mitral stenosis and insufficiency, and aortic insufficiency. In children, mitral insufficiency remains the major sequela of rheumatic heart disease.
DIAGNOSIS
Diagnosis depends on recognition of one or more of the classic symptoms (carditis, rheumatic fever without carditis, polyarthritis, chorea, erythema marginatum, or subcutaneous nodules) and a detailed patient history. Laboratory data support the diagnosis:
White blood cell count and erythrocyte sedimentation rate may be elevated (during the acute phase); blood studies show slight anemia due to suppressed erythropoiesis during inflammation.
C-reactive protein is positive (especially during acute phase).
Cardiac enzyme levels may be increased in severe carditis.
Antistreptolysin-O titer is elevated in 95% of patients within 2 months of onset.
Electrocardiogram changes aren’t diagnostic, but PR interval is prolonged in 20% of patients.
Chest X-rays show normal heart size (except with myocarditis, heart failure, or pericardial effusion).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


