Overview
Diseases of the heart fit into several general categories: congenital heart disease, ischemic heart disease, valvular diseases, and diseases of the myocardium (i.e., cardiomyopathies). Pericardial diseases and cardiac tumors are an additional small subset of conditions affecting the heart. A common manifestation of many different forms of heart disease is congestive heart failure (CHF). In general terms, congestive heart failure is the inability of the heart to pump enough blood to supply the body’s oxygen requirements. It can represent failure of cellular adaptation (e.g., decompensated hypertrophy due to hypertension or chamber dilation due to regurgitant valves) or the outcome of myocardial damage caused by other diseases (e.g., scarring due to ischemic injury, inflammation, or accumulation of iron in hemochromatosis).
Classic symptoms of heart disease are chest pain or discomfort, dyspnea (including orthopnea and paroxysmal nocturnal dyspnea), palpitations, syncope, and edema. Dyspnea is an uncomfortable awareness of breathing. Orthopnea is dyspnea when in the recumbent position due to increased venous return and increased pulmonary venous pressure. Patients with orthopnea sleep upright on pillows to avoid becoming short of breath. Paroxysmal nocturnal dyspnea is when patients awaken with dyspnea 2–4 hours after falling asleep (due to central redistribution of peripheral edema).
An understanding of heart sounds is important in the clinical evaluation of heart disease. The S1 sound is caused by closing of the mitral and tricuspid valves, and the S2 sound is caused by closing of the aortic and pulmonary valves. In a patient with hypertension (systemic or pulmonary), closing of the associated valve (aortic or pulmonic) is accentuated (louder); in a patient with stenosis, the closing is diminished in strength (softer sound). S2 is physiologically split during inspiration (aortic, A2, first and pulmonic, P2 second)—increased venous return to the right side of the heart delays closure of the pulmonic valve and decreased return to the left side speeds closure of the aortic valve. Wide splitting of S2 is caused by a greater than normal delay in pulmonic closure (e.g., right bundle branch block, pulmonic stenosis) or earlier aortic valve closure due to decreased left ventricular volume (e.g., mitral regurgitation, ventricular septal defect). Paradoxical splitting (P2 first and A2 second) occurs with delayed closure of the aortic valve (e.g., left bundle branch block, aortic stenosis). A pathologic S3 occurs with ventricular systolic dysfunction during the rapid filling phase of diastole or from impact of the left ventricle against the chest wall. It is particularly common in the setting of CHF. S4 is from ejection of blood from the atrium into a noncompliant ventricle, as might be encountered in the setting of ventricular hypertrophy related to systemic hypertension, or in the setting of an acute myocardial infarct.
Congenital Heart Disease
Overview: There are three main categories of congenital heart disease: conditions causing a right-to-left shunt; conditions causing a left-to-right shunt; and conditions causing obstruction. In a right–to–left shunt, deoxygenated blood from the right side of the heart goes to the left side; thus, deoxygenated blood is delivered to the body. This type of shunt usually results in cyanosis at the time of birth. A left–to–right shunt increases the amount of blood delivered to the right side of the heart and will result in hypertrophy and dilation of the right atrium or right ventricle (or both), depending upon the type of shunt. Eventually, the pressure in the right side of the heart increases and surpasses that in the left side of the heart, resulting in a reversal of the shunt from left-to-right to a right-to-left shunt. This change is called Eisenmenger syndrome. With obstruction, an abnormally formed valve or vessel leads to pressure overload of the involved atrium or ventricle.
Causes of right-to-left shunt
Tetralogy of Fallot
- Morphology of tetralogy of Fallot
- Pulmonary stenosis.
- Right ventricular hypertrophy (as a result of the pulmonary stenosis).
- A ventricular septal defect shunts blood to the left side of the heart.
- The aorta overrides the ventricular septal defect.
- Pulmonary stenosis.
- Important points: Tetralogy of Fallot is the most common cause of a right-to-left shunt in newborns.
- Complications of tetralogy of Fallot: Erythrocytosis, paradoxical emboli (through ventricular septal defect), endocarditis, and ventricular arrhythmias.
- Clinical presentation of tetralogy of Fallot: Cyanosis and failure to thrive; patients squat to alleviate symptoms by increasing venous return. The classic pattern seen on chest radiograph is the boot-shaped heart. Patients often have paroxysms of cyanosis (“tet fits”) associated with states of increased cardiac output that increase right-to-left shunting such as crying, exercise, and hot baths.
Tricuspid atresia: The gross morphology of tricuspid atresia is an atretic tricuspid valve that obstructs the flow of blood from the right atrium to the right ventricle. A ventricular septal defect or atrial septal defect is usually present. Approximately 75% of infants with tricuspid atresia are cyanotic within the first week of life.
Truncus arteriosus: The gross morphology of truncus arteriosus is caused by abnormal development of the ventricular outflow tracts, resulting in the presence of a common arterial conduit that receives a mixture of blood from the left and right ventricles. These patients present with symptoms initially due to left-to-right shunting, and they become cyanotic within 3 to 4 months because of progressive pulmonary vascular resistance.
Totally anomalous pulmonary venous return: The gross morphology is that the pulmonary veins do not return to the left atrium as normal, but rather drain abnormally into a left innominate vein or coronary sinus. Pulmonary venous blood ultimately reaches the left atrium through an atrial septal defect or patent foramen ovale.
Transposition of the great vessels: The gross morphology is that the aorta arises from the right ventricle and the pulmonary trunk arises from the left ventricle, producing two completely separate circuits. One circuit is the right ventricle → body → right atrium, and the other is the left ventricle → lungs → left atrium. Transposition of the great vessels must be combined with a ventricular septal defect, patent ductus arteriosus, or patent foramen ovale, which the patient needs to mix blood for any survival period to be possible after delivery.
Causes of left-to-right shunt
Ventricular septal defect (VSD)
Gross morphology: Defect in the interventricular septum. Most VSDs are in the membranous septum (90%). Many close spontaneously, and particularly those limited to the muscular septum (Figures 10-1 and 10-2).
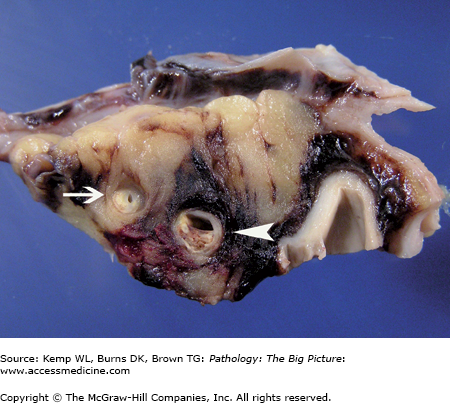
Figure 10-1.
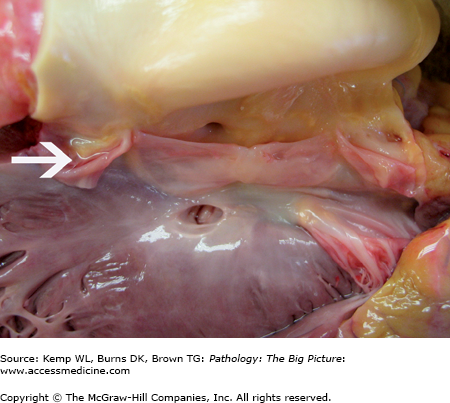
Ventricular septal defect and bicuspid aortic valve. The ventricular septal defect is in the membranous septum. The bicuspid aortic valve has one large cusp (above the membranous ventricular septal defect) with a midline raphe and a smaller cusp. During dissection, the smaller cusp was divided (arrow). Courtesy of Dr. Sheila Spotswood, Dallas County Medical Examiner’s Office, Dallas, TX.
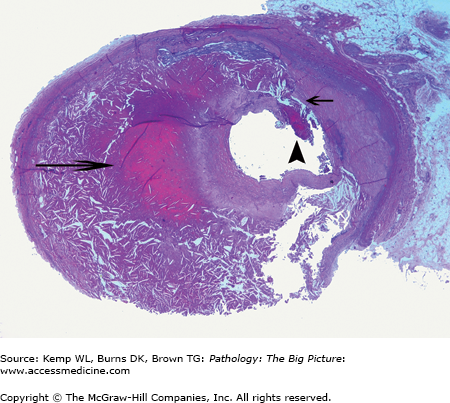
Figure 10-2.
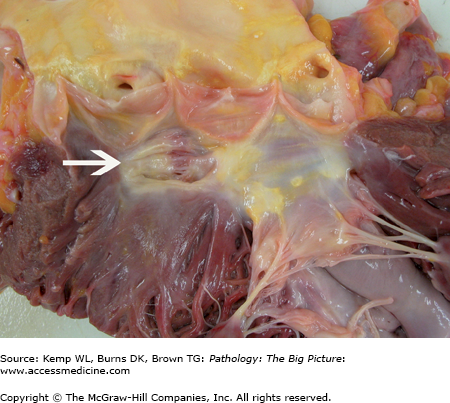
Healed ventricular septal defect. This patient had a membranous ventricular septal defect, which, like most, spontaneously closed. In this case, closure was due to the anterior leaflet of the tricuspid valve adhering to the region of the defect. This view shows the closed ventricular septal defect from the aortic aspect (arrow).
Incidence: VSD is present in 30–60% of all patients with a congenital heart defect, making it the most common congenital heart defect and the most common cause of a left-to-right shunt.
Complications of VSDs
- Endocarditis
- Left-to-right shunt with increased flow of blood to the right ventricle, leading to hypertrophy and dilation and eventually to Eisenmenger syndrome (see above).
- Paradoxical embolus: An embolus passes into the right ventricle and then into the left ventricle through the VSD. From the left ventricle, the embolus travels via the aorta to the systemic arteries.
Clinical presentation of VSD: Wide physiologic splitting of S2 and holosystolic murmur. Spontaneous closure of VSD occurs in up to 50% of patients.
Atrial septal defect (ASD)
Gross morphology: Defect in the interatrial septum.
Three types of ASDs based upon location
- At the fossa ovalis (secundum-type): Most common (Figure 10-3).
- Below the fossa ovalis (primum-type): Second most common.
- Near the superior vena cava (sinus venosus type): Rare.
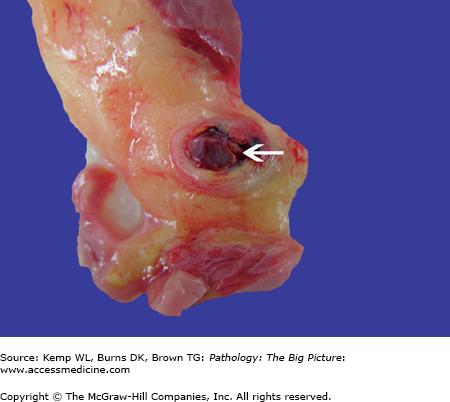
Figure 10-3.
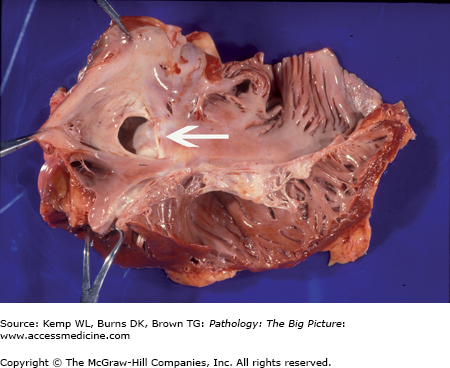
Atrial septal defect, secundum-type. This 32-year-old patient had an undiagnosed secundum-type atrial septal defect (arrow). The right atrium is also dilated, as would be expected in a patient with a long-standing unrepaired atrial septal defect (from increased blood flow to the right side of the heart).
Incidence: Second most common cause of left-to-right shunt. Up to 50% of all patients with congenital heart defects have an ASD.
Complications of ASDs
- Left-to-right shunt with increased flow of blood to the right atrium. Secondary pulmonary hypertension is less common than with VSDs because of the low pressure flow of the left-to-right shunt.
- Paradoxical embolus.
- Endocarditis.
Clinical presentation of ASDs: Widely split and fixed S2 because of increased venous pressure and delayed closure of pulmonic valve.
Patent ductus arteriosus (PDA)
Gross morphology: Retention of the patency of the duct between the aorta and pulmonary trunk. The ductus arteriosus, along with the foramen ovale, shunts blood around the lungs in the fetus because the lungs are not needed to oxygenate the blood during fetal development.
Clinical presentation of PDA
- In the normal neonate, spontaneous closure of the ductus arteriosus in response to an increase in PaO2 associated with breathing normally occurs within the first 12 hours of life.
- The classic sign of a persistent PDA is a continuous “machine-like” murmur.
- Use of prostaglandin E will keep the shunt open. Use of indomethacin will close a PDA.
Atrioventricular septal defect
Types of atrioventricular septal defects
- Partial: Primum atrial septal defect combined with cleft mitral valve.
- Complete: Common atrioventricular canal.
Important point: Associated with Down syndrome.
Causes of obstruction as pathophysiologic mechanism of congenital heart disease
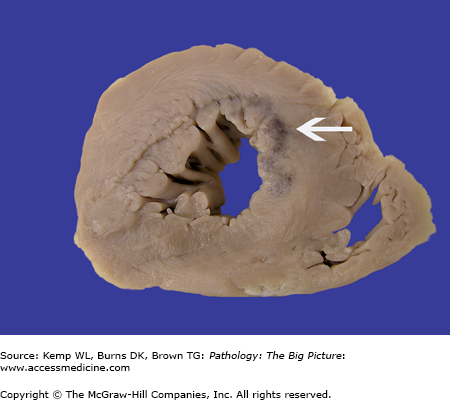
Figure 10-4.
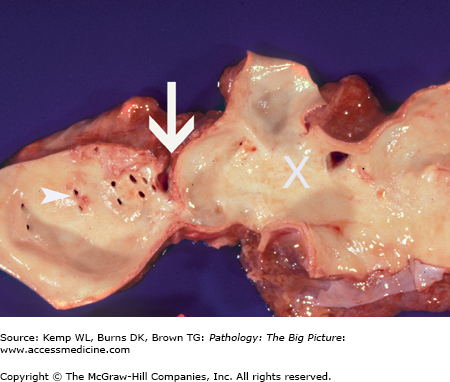
Coarctation of the aorta. This 40-year-old man had an undiagnosed coarctation of the aorta (arrow). For orientation, the arrowheads indicate ostia of the intercostal arteries, and the large white X marks the site of the aortic arch. Associated with this coarctation was a true bicuspid valve (i.e., two cusps of equal size). The patient also had cardiac hypertrophy caused by pressure overload induced by the coarctation of the aorta.
Types of aortic coarctation (based upon location in relationship to the ligamentum arteriosum)
- Preductal: Present with symptoms early; identified in infancy.
- Postductal: Present with symptoms later; identified in early adulthood.
- The presence or absence of a PDA is also important in the classification of a coarctation of the aorta.
Conditions associated with coarctation of the aorta
- About 25% of patients with coarctation of the aorta have a bicuspid aortic valve.
- Intracranial saccular (berry) aneurysms.
- Coarctation of the aorta is present in up to one third of patients with Turner syndrome.
Clinical presentation of coarctation of the aorta
- In coarctation of the aorta associated with a PDA, patients have cyanosis at birth, usually of the lower half of the body.
- In coarctation of the aorta without an associated PDA, patients present with systemic hypertension with lower blood pressure in the arms than in the legs. Notching of ribs is seen on chest radiograph; the notching is caused by dilated collateral vessels supplying the lower part of the body.
Ischemic Heart Disease
Overview: The term ischemic heart disease refers to inadequate perfusion of the myocardium, most commonly due to atherosclerosis of the coronary arteries. Ischemia causes functional disturbances of the heart, including (1) impaired relaxation, which occurs first, causing diastolic dysfunction; (2) impaired contraction, which occurs second, causing systolic dysfunction; (3) myocardial stunning, a prolonged (hours to days) but reversible dysfunction after an acute ischemic event; and (4) myocardial hibernation, which occurs when oxygenation is adequate to maintain viability of the myocardium but cannot support normal function. If the ischemia lasts long enough, death of myocardial cells ensues.
Clinically, decreased blood flow to the myocardium causes chest pain (angina). The nature of the obstruction to blood flow determines the disease type and outcome. A fixed obstruction (70% or greater stenosis) will cause ischemia of the myocardium when the demand for oxygen is increased (such as during exercise), but at rest, flow is adequate (therefore there is no pain when the patient is at rest). Chest pain due to a fixed obstruction is called stable angina.
Myocardial necrosis does not occur during classic episodes of stable angina. In a patient with coronary atherosclerosis, changes in the coronary artery plaque morphology, such as rupture or hemorrhage, may occur suddenly and may cause acute myocardial ischemia; if prolonged, such acute ischemic episodes may cause necrosis (myocardial infarction). Unstable angina, non–STEMI (non-ST elevation myocardial infarct), and STEMI are clinical terms that designate, in increasing order of severity, possible consequences of a change in the plaque morphology, depending upon the length of obstruction. The signs of myocardial ischemia include ST depression on the electrocardiogram (ECG) and an S4 on physical examination, with the S4 due to decreased ventricular compliance.
Overview: The four clinical syndromes are stable angina, unstable angina, variant angina, and myocardial infarct, the first three of which will be described below. Unstable angina and myocardial infarcts (both non-STEMI and STEMI) can be further subdivided as acute coronary syndromes, which will be described after this section.
Stable angina
Basic description: Chest pain due to fixed obstruction of the coronary arteries. The chest pain occurs predictably with exercise and ceases with rest.
Mechanism of stable angina: Stable angina is produced by a fixed plaque causing stenosis of 70% or more of the vessel lumen (Figures 10-5 and 10-6). At rest, the heart can get a sufficient amount of blood around a site with this degree of stenosis to supply the heart (there is no ischemia; therefore, there is no chest pain). With exercise, myocardial oxygen requirements increase, but the heart cannot pump enough blood past the blockage to meet the metabolic demands of the myocytes. The myocytes become ischemic and chest pain occurs. With increased myocardial oxygen requirements, normal coronary vessels dilate. Atherosclerotic vessels are unable to dilate appropriately in this setting, however. With cessation of exercise, myocardial needs decrease and ischemia stops. The symptoms are consistent (i.e., a set amount of exercise triggers the chest pain in a predictable fashion).
Figure 10-5.
Atherosclerosis seen in a cross-section of the proximal left anterior descending coronary removed from the heart. This patient has mild to moderate atherosclerosis of the coronary arteries. The vessel at the arrowhead is about 50–60% stenotic. This patient would not be expected to have chest pain associated with this lesion. The vessel at the tip of the arrow is about 75–85% stenotic. This patient would be expected to have stable angina associated with this lesion.
Figure 10-6.

Atherosclerosis of the coronary artery. The sections at the arrows have mild to moderate atherosclerosis, with approximately 55–70% stenosis of the lumen. These changes would not likely cause chest pain. The vessel at the arrowhead has severe stenosis due to atherosclerosis (90–95%), with only a slit-like vessel lumen remaining. This patient would be expected to have, at the least, stable angina.
Clinical presentation of stable angina
- Description of pain: The chest pain is described as squeezing or pressure, such as a heavy weight.
- Location: Retrosternal; commonly mistaken for “indigestion.”
- Radiation of pain: To the neck, epigastrium, jaw, and ulnar aspect of the left arm.
- Duration of pain: Less than 2 to 10 minutes.
- Context: Triggered by stress, either physical or emotional. Generally alleviated by rest or nitroglycerin.
- Other symptoms occurring along with chest pain: Nausea and vomiting, diaphoresis, shortness of breath, and fatigue. In older patients and diabetics, shortness of breath may be the only symptom of angina.
Unstable angina
Basic description: Chest pain that falls into one of several patterns; namely, chest pain that is more severe, prolonged, or frequent in a patient with previous stable angina (“crescendo” angina); chest pain occurring at rest or with minimal exertion; or chest pain of new onset associated with minimal exertion.
General mechanism of unstable angina: Unstable angina is produced by a change in the plaque. Usually the change is acute (Figure 10-7), although a stable plaque causing 90% or greater stenosis could cause angina when at rest. In unstable angina, acute changes in plaque morphology abruptly deprive the myocytes of sufficient oxygen and ischemia occurs. Unstable angina has also been called preinfarction angina, because this form of chest pain is often a prelude to a myocardial infarct. Ischemia leads to infarction if the precipitating event is not corrected.
Figure 10-7.
Intraplaque hemorrhage with resultant rupture of plaque and thrombus formation. This patient had a hemorrhage within an atherosclerotic plaque (long arrow) that has caused rupture of the thin fibrous cap (short arrow) and thrombus formation (arrowhead). This patient would be expected to have unstable angina (because of the recent change in the plaque), which may progress to frank myocardial infarction, depending upon the degree and duration of occlusion. Hematoxylin and eosin, 40×.
Specific mechanisms of plaque change: Atherosclerotic plaques with a fairly thin fibrous cap are more likely to develop acute changes than those with a thick fibrous cap. Acute plaque change can be due to hemorrhage into the plaque, rupture of the plaque, or erosion or ulceration of the plaque.
- Hemorrhage into the plaque: Plaques have blood vessels. Hemorrhage can lead to an increase in the size of the plaque, with resultant increased stenosis of the vessel, or the hemorrhage can lead to rupture of the fibrous cap and intraluminal thrombus formation.
- Rupture of the plaque with resultant intraluminal mural thrombus.
- Erosion or ulcer: The fibrous cap can erode or ulcerate. This change results in hemorrhage and sometimes exposure of the atheromatous core, which can result in intraluminal thrombus formation.
Cellular basis of plaque disruption: Cells die, releasing lipid. Activated macrophages and mast cells release metalloproteinase enzymes, which break down matrix proteins. T lymphocytes in the plaque produce cytokines, which block production of collagen. Other factors, such as mechanical and hemodynamic stresses, may also contribute to plaque disruption.
Clinical presentation of unstable angina: Characteristic symptoms of unstable angina are the same as stable angina, but duration may be longer and chest pain occurs with rest or increasing frequency. (Refer to the basic description and mechanism of unstable angina.) All new onset cases of angina are considered to be unstable angina until proven otherwise.
Variant angina
Basic description: Angina occurring unpredictably with rest. There are two forms: Prinzmetal angina and pure vasospastic angina.
- Prinzmetal angina: Dynamic (vasospastic) angina that occurs at the site of an existing atherosclerotic plaque. It often occurs at rest, and may be triggered by use of β blockers or hyperventilation. Unlike classic angina, which is associated with ST depression, Prinzmetal angina is characterized by a profound ST elevation, although progression to myocardial infarction is rare. Prinzmetal angina usually responds to nitrates or calcium channel blockers.
- Pure vasospastic angina: Associated with normal coronary arteries (i.e., no atherosclerosis).
Mechanism of variant angina: Coronary artery vasospasm.
Complications of variant angina: Can cause ischemia and, therefore, precipitate ventricular arrhythmias and sudden cardiac death. Uncommonly, myocardial infarcts may occur.
Acute Coronary Syndromes
Myocardial Infarcts
Basic description: Death of a segment of myocardium due to prolonged ischemia, usually due to obstruction of a coronary artery.
- STEMI (previously called transmural or Q wave infarct): The full thickness of the wall of the ventricle is affected. The mechanism of an STEMI infarct is complete occlusion of the coronary artery (100% stenosis with no blood flow) for more than 2–4 hours, which is the length of time required for all the myocytes throughout the entire thickness of the wall to die (Figure 10-8).
- NSTEMI (previously called subendocardial or non–Q wave infarct): The full thickness of the wall is not affected; only the subendocardial myocytes are affected (Figure 10-9). The mechanism of NSTEMIs is deprivation of the wall of the ventricle of oxygen for enough time to cause necrosis, but not enough time to cause full thickness necrosis. Since the last myocytes to receive blood are those nearest the endocardium and because blood comes into the wall from the epicardial surface, subendocardial myocytes are the most prone to injury from decreased blood flow. NSTEMIs of this region (i.e., subendocardial in location) almost always result from one of three conditions.
Figure 10-8.
Minimal coronary artery atherosclerosis with acute thrombus. This patient had mild coronary artery atherosclerosis. The thin rim of yellow is the plaque, which causes no more than 10% stenosis of the lumen of the vessel. Yet, despite its small size, the plaque ruptured and an occlusive thrombus formed (arrow). The patient most likely will develop an ST elevation myocardial infarct (STEMI), with full thickness involvement of the myocardium in the area supplied by the vessel.
Complete occlusion of the vessel by a thrombus, which lyses prior to full development of a transmural infarct (i.e., the thrombus breaks up before 2 hours).
A sudden change in the plaque occurs, which significantly narrows the lumen but does not completely occlude it. For example, if stenosis rapidly goes from 50% to 90%.
Generalized poor perfusion of the heart because of shock.
- ECG changes: Both cause ST depression; Q waves do not develop.
- Cardiac enzyme testing: Unstable angina does not cause an elevation in cardiac enzymes (see clinical diagnosis of STEMI infarct). NSTEMI infarcts do cause an elevation in cardiac enzymes.
- Clinical presentation: Symptoms include retrosternal, substernal, or epigastric chest discomfort, which can radiate to the arm. This chest discomfort is often associated with dyspnea, nausea, and/or diaphoresis.
- Unstable angina and NSTEMI can present with similar clinical and ECG findings. Enzyme testing will distinguish the two (see clinical diagnosis of STEMI infarct).
- About 15% of patients with unstable angina will progress to myocardial infarction.
Basic description: Death of cardiac myocytes releases enzymes into the blood (e.g., CK-MB and troponin I), which can then be detected.
Monitoring CK-MB and troponin I
CK-MB: Isoform of creatine kinase (other isoforms include –MM and –BB). In the heart, most of the CK is CK-MB; therefore, if the ratio of CK-MB to total CK is high, it is consistent with damage to the heart. The level of CK-MB rises quickly after a myocardial infarct (within 4–8 hours), peaking at 24 hours, and falls to a normal level within 3 days.
Troponin I: Very specific for damage to the heart. Troponin I rises within 3–4 hours after a myocardial infarct and remains elevated for 10–14 days.
ECG findings of STEMI infarct
- Myocardial infarction: Focal ST elevation.
- Q-waves: Infarcted myocardium cannot conduct electrical forces; therefore, the electrical forces are directed away from the electrode and a Q wave results.
Morphology of a myocardial infarct based upon age (Table 10-1)
- Microscopic morphology: At 4 to 12 hours, coagulative necrosis begins; at 12 hours to 3 days, infiltration of neutrophils begins and grows more prominent; at 3 to 7 days, macrophages and neutrophils are present; at 7 to 10 days, granulation tissue develops; at 10 days or more, the start of collagen deposition; and at 8 weeks or more, dense fibrous scar is present.
- Gross morphology: Prior to 12 hours of age, unable to see myocardial infarct; at 1 to 3 days, neutrophil infiltrate causes yellow discoloration; at 3 to 7 days, macrophages engulfing dead cells cause shrinkage of tissue; and by 8 weeks, only a dense scar is present (Figures 10-10

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


