- The story of anticancer therapeutics begins during the First World War, when bone marrow suppression was observed in soldiers who had been killed by mustard gas.
- In 1942, a patient in the terminal stages of lymphosarcoma was successfully treated by the administration of a chemical, the nitrogen mustard tris(β-chloroethyl)amine.
- The discovery of methotrexate (an antimetabolite of folic acid) in the 1950s heralded the development of the antimetabolite class of anticancer agents.
- The more complete description of the molecular pathoetiology of cancers that has evolved in the last 20 years has identified multiple new potential targets for therapeutic intervention, as exemplified by the development of tyrosine kinase inhibitors such as imatinib.
- The drug discovery pipeline consists of the following steps:
- Choosing a cancer-relevant “druggable” target;
- Finding a series of chemical entities or biomolecules (hits) that are able to modulate that target by screening in vitro;
- Finding the lead agent of the series;
- Optimizing the lead candidate; and
- Developing the candidate for clinical trials.
- Choosing a cancer-relevant “druggable” target;
- Medicinal chemists have exploited a variety of sources to find leads, including: natural products (e.g. taxol), the application of pharmacological tools such as screening compound libraries in vitro (e.g. vatalanib) and in silico (e.g. HA14-1), established drugs (for “me betters,” such as carboplatin), competitor patents, publications, and even serendipity (e.g. cisplatin).
- In addition to developing traditional compounds, new therapeutic approaches are needed in anticancer drug research, including wider exploration of metal- and metalloid-based compounds, vaccines, nucleic acid–based therapies (e.g. RNAi), and utilization of new techniques such as photodynamic therapy (PDT).
- The lead-development phase focuses on the metabolism and distribution of the drug in the body, on its efficacy against the target (pharmacokinetic (PK) and pharmacodynamics (PD) studies), and on the avoidance of toxicity. This may result in the development of a pro-drug that allows the active drug to form following metabolism in the body. The pro-drug approach has also been exploited so that the pro-drug is activated only near the tumor (staying innocuous in the rest of the individual to diminish off-target effects).
- The pre-clinical lead-development phase involves collating all necessary information required for a drug to enter clinical trials. Bioavailability, toxicity, safety data, manufacturing information, and other regulatory documentation are submitted to various regulatory agencies for ethical and scientific approval. Approval then allows the potential drug to enter clinical trials (see Chapter 16 on cancer clinical trials).
Introduction
Chemotherapy is defined as the use of chemicals to treat diseases, but it has become synonymous with cancer drug therapy. The key cellular processes involved in the pathoetiology and behavior of cancer cells have been outlined in other chapters of this book. Not surprisingly, many of these are or will become the target of anticancer drugs aiming to cure, arrest, or prevent cancer, on the one hand, or to prevent critical life-shortening behaviors such as invasion or metastases, on the other; Chapter 18 will provide an overview of those drugs already in clinical use. This chapter describes the processes by which these and future cancer drugs originate, and Chapter 16 concentrates on biologically targeted therapies and clinical trials. What will readily become apparent is that for a new compound to be developed and to successfully pass through the various stages described here is nothing short of miraculous. The route to the next blockbuster drug is strewn with the remains of tens of thousands of compounds which have failed on the way.
We describe here the various stages that constitute the general modern approach to drug discovery. Each developmental stage will be addressed with examples of specific therapeutics or potential future drugs. Where this chapter ends, the next on cancer clinical trials begins. The focus here is specifically on drugs to treat cancer. The development of vaccines to prevent infection with potentially tumor-causing viruses such as HPV, and drugs to assist smoking cessation, despite their potential value in preventing cancers, are not discussed.
A cautionary note – cancer cells have a nasty habit of treating cancer therapy as another opportunity for driving natural selection (or, indeed, even of accelerating their acquisition of potentially advantageous mutations). The net result is that cancers rapidly and often inevitably acquire resistance to the effects of drugs that target specific cancer pathways (see Chapter 5). A major task in the future of cancer drug development will be to overcome this using strategic combinations of agents and dosing schedules, or to target critical and nonredundant targets, such as c-Myc, where no “escape route” may be available.
Historical Perspective
Cancer has been known for most of recorded history (Appendix 1.1), yet at the start of the 20th century there was not a single convincing report of a medical (nonsurgical) cure of cancer.
Ironically, the first steps toward medicines for cancer stem from an inadvertent consequence of the use of mustard gas as a chemical weapon during the First World War (1914–18). In 1919, Krumbhaar published results of hematological examinations of patients in a base hospital in France who had been poisoned by mustard gas (sulfur mustard). Direct toxicity to the bone marrow, which peaked at 2 weeks after exposure, coincided with the highest mortality. It is interesting to note with hindsight how common this finding is as an unwanted side effect of so many chemotherapeutic agents.
In the United Kingdom during the late 1920s and early 1930s, Beremblum was investigating the effects of mustard gas on tar-triggered tumors in mice (tar was a known carcinogen), working on the then-plausible hypothesis that mustard gas would increase the carcinogenic effects of tar by inducing hyperemia. Somewhat to his surprise, tumors were inhibited in these mice, and by serendipity he had identified a potential anticancer compound.
James Ewing had been “impressed by the peculiar and specific nature of mustard-gas burns” while serving at the US Army Medical Museum, an observation that prompted Adair and Bagg to study effects of mustard gas on cancer-bearing animals and human cancer patients. In 1931 they published the regression effects of mustard gas (when topically administrated in alcohol solution) on several types of superficial tumors in 12 patients of the Memorial Hospital in New York. Although the nitrogen mustards were not a definite cure for cancers and the tumor regression was temporary, this was “proof-of-concept,” and the search for agents that would be more selective toward specific types of malignancies began. Thus, the combination of “wartime” ingenuity, coupled with the application of suitable test systems such as animal models, opened up the era of cancer drug development.
Importantly, in the mid-1930s Alexander Haddow had published in Nature the paradoxical observation that three polycyclic hydrocarbons known as carcinogens, chemical substances that induced cancer in animal models, could also retard tumor growth in rats (Haddow’s paradox), whilst noncarcinogenic but similar chemicals had no effect in delaying tumor growth (see Chapter 10 for a discussion about DNA damage as a cause and cure for cancer).
In the late 1930s Carl Voegtlin, the first director of the National Cancer Institute (NCI) of the National Institutes of Health in the United States, in a series of lectures on chemotherapy emphasized how little was known about the mode of action of chemicals in humans and, most importantly, the need to find chemical treatments for cancer, given the incomplete achievements of surgical and radiation treatments.
Nitrogen Mustards
Regarding the discovery of DNA-damaging agents, it can be argued that the formal history of chemotherapy began in the 1940s, when nitrogen mustards were used for the treatment of lymphoma.
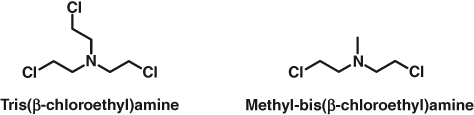
Goodman and Gilman, who will need no introduction to students of pharmacology, were recruited to test a series of chemical warfare agents developed earlier in the 20th century for their potential therapeutic value (a contract was signed by Yale University and the Office of Scientific Research and Development – an agency of the US government created in 1941 to manage scientific research for military purposes during World War II). They were joined on the project by Philips and Allen. Later, in the early 1960s, in narrating the events that led to the very first clinical trial in humans of nitrogen mustards, the chemical entities tris(β-chloroethyl)amine and methyl-bis(β-chloroethyl)amine, Gilman explained,
Close contact was maintained between investigators by means of circulated research reports and frequent meetings. This accounted for the rapid elucidation of the unique and fascinating properties of the nitrogen mustards. Contrary to the present opinion of many, perhaps no compound had been more thoroughly studied prior to clinical trial than were the nitrogen mustards. The point to be emphasized is the collaborative nature of the basic investigations on the nitrogen mustards which led to their clinical trial.
The rapid progress of the chemists involved in the research shed light on the biotransformation of the nitrogen mustards. Scheme 15.1 shows the cyclization of β-chloroethylamines and the formation of the highly reactive aziridinium ring. In the case of tris and bis(β-chloroethyl)amines, this involves the formation and reaction of successive aziridinium rings. Much information was collected about the distribution, pharmacodynamics, and toxicity of the agent in animal models.
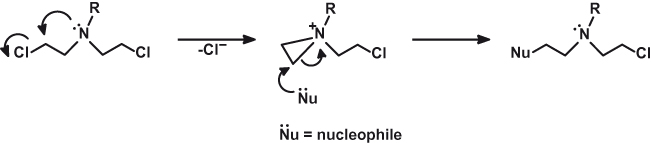
Finally, Gilman, Goodman, and Philips took the anatomist Dougherty on board, and efficacy experiments on lymphoma in mice were carried out. Strikingly positive results from this group convinced Gustav Lindskog (a thoracic surgeon) to treat a nonresponding X-ray patient in the terminal stages of lymphosarcoma with tris(β-chloroethyl)amine (Fig. 15.1). This was in December 1942. The results, subject to a wartime secrets policy, were not published until 1946, when the restrictions were lifted. The same reason was behind the publication delay of independent but similar studies by Wilkinson and Fletcher in the United Kingdom. The authors tried tris(β-chlorethyl)amine hydrochloride and methyl-bis(β-chlorethyl)amine hydrochloride (Fig. 15.1) in 18 patients with one of the following malignancies: chronic myeloid and lymphatic leukemias, Hodgkin disease, or polycythemia. The level of response varied widely, with the best responses observed in chronic myeloid leukemia and Hodgkin disease. Today we know that the mechanism of action of nitrogen mustards involves alkylation of DNA (Scheme 15.1, where the nucleophile is DNA), which ultimately disrupts cell proliferation and leads to cell death.
These studies highlighted the importance of interdisciplinary collaboration and also suggested that finding a single “magic bullet” for the treatment of all cancers, even of the same type, might be unattainable.
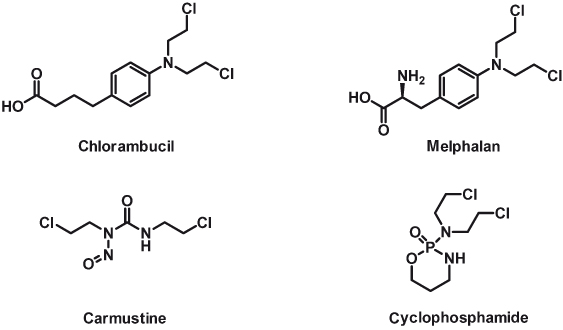
As a result of these discoveries, other types of alkylating agents derived from the original nitrogen mustards were developed. Many of these are in clinical practice today (Fig. 15.2), most notably chlorambucil, melphalan, carmustine, and cyclophosphamide (discussed in this chapter).
Methotrexate
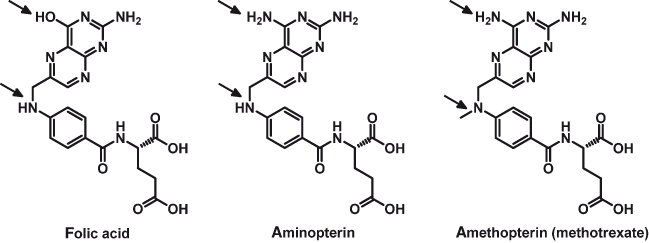
The antimetabolites: One of the earliest examples of hypothesis-led drug development resulted in the trial of an anti–folic acid metabolite by Sidney Farber and colleagues. Following the identification of folic acid as a requirement for leukemia development, they decided to administer an analogue of folate, which could compete with folate binding to the biological target. This became known as antimetabolite therapy, since the analogue of folate is an antimetabolite (a chemical entity similar enough to a natural metabolite to mimic it in a normally occurring biochemical reaction in the cell, but different enough to alter the cell’s normal function). An antimetabolite drug inhibits a normal metabolic process involved in causing disease. They first tested the analogous compound, aminopterin (Fig. 15.3), which differs in structure from folate in that an OH group is replaced with an NH2 group. Sixteen infants and children with acute leukemia were treated intramuscularly, and a marked improvement (manifested clinically, histologically, and pathologically) occurred in 10. The severe toxicity of the drug drove them toward the discovery of more efficient and less toxic analogues, such as amethopterin, now known as methotrexate (Fig. 15.3). Methotrexate differs from folate in having the same NH2 group as aminopterin and, additionally, replacement of a hydrogen atom in the NH group by a methyl group (CH3). Its clinical development was favored as it had a better therapeutic window. Today, methotrexate is still used in the clinic, and we know that it competitively inhibits dihydrofolate reductase (DHFR) at picomolar concentrations. DHFR catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF), an essential cofactor in the biosynthesis of thymidylate monophosphate (dTMP). Inhibition of DHFR leads to a deficiency of dTMP as DHF cannot be recycled, which ultimately causes cell death.
Figure 15.3 Structures of folic acid and its antimetabolites aminopterin and amethopterin (methotrexate). The arrows show the sites of the structural modifications.
6-Mercaptopurine
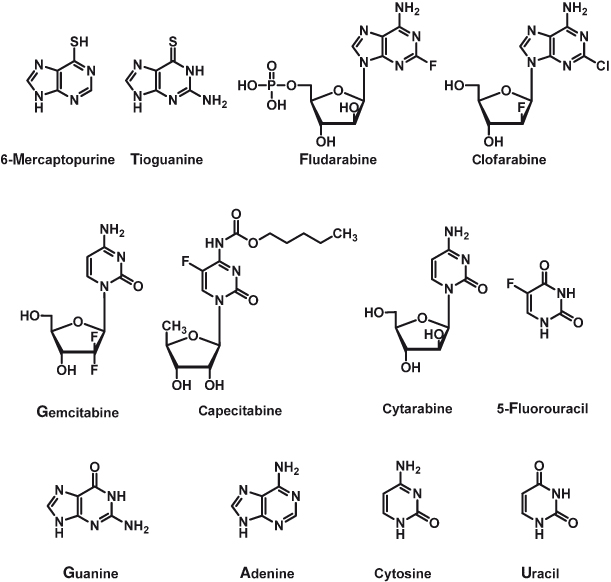
Burchenal, an oncologist based in the Sloan-Kettering Institute in New York who had shown interest in the effects of methotrexate, extended the rational of antimetabolite therapy to nucleobases in the hope of disturbing nucleic acid synthesis. Burchenal established a collaboration with pharmaceutical chemist Hitchings and colleagues who had been investigating the relationship between the chemical structures for derivatives of purines and pyrimidines and their interference with the biosynthesis of nucleic acids and metabolism in bacteria. The discovery of the anti-leukemia activity of 6-mercaptopurine (Fig. 15.4) is attributed to this collaboration. This extensive work was reported in a landmark publication in the journal Blood in 1953. Sixty years on, the concept is still alive, and a number of antimetabolites of nucleic acid building blocks are now used in the clinic against different types of cancer. Examples are tioguanine, fludarabine, clofarabine, gemcitabine, capecitabine, cytarabine, and 5-fluorouracil (Fig. 15.4).
Figure 15.4 Structures of some DNA alkylating agents and the four nucleobases to which their structures relate.
The toxicity of nitrogen mustards slowed their adoption into clinical trials. However, in the 1950s, thinking had begun to change, and both the public and researchers became interested in finding drugs that can affect cancer. The Developmental Therapeutics Program (DTP; originally called the Cancer Chemotherapy National Service Center (CCNSC)) was created in the United States in 1955 as a drug discovery and development arm of the National Cancer Institute (NCI). There was also a growing understanding that there are many types of cancers and there is no panacea for cancer therapeutics. In the 1960s, new strategies evolved using “drug cocktails.” As seen in Chapter 18, drug combinations are now commonplace and responsible for much of the current clinical successes of chemotherapy for cancer.
The crucial early role of not-for-profit organizations, academia, and governments should not be overlooked – there was little incentive for the pharmaceutical industry (motivated primarily by the need to be profitable) at that time when the risk was highest, the need for investment was greatest, and the potential market was the most uncertain – a situation completely reversed now by the longevity of many cancer patients and the near-ubiquitous application of cancer drugs to those patients. However, a government-sponsored program allowed academic investigators to have access to resources only accessible to large pharmaceutical firms. The NCI and its Developmental Therapeutics Program played a key role in the development of anticancer drugs, in particular after the National Cancer Act in 1971, when President Richard Nixon declared “war on cancer” and the NCI was given a new mission: to support research in a way in which basic discoveries translate into actual applications to truly reduce cancer incidence, morbidity, and mortality.
The second half of the 20th century changed views on cancer research for pharmaceutical companies. Discoveries with promising turnovers such as taxol or cisplatin did indeed spark a newly found interest in the pursuit of anticancer drugs. Cancer drug development has since transformed from a government- and charity-supported, low-budget research to a multibillion-dollar industry. Additionally, targeted therapies have driven a highly competitive race to bring ever-newer tyrosine kinase inhibitors and other targeted agents into clinical trials in unprecedented numbers.
The Drug Discovery Process and Preclinical Development of a Drug
It has been estimated that the discovery of a drug and its preclinical development take about 12−15 years and cost $0.8−1.7 billion. The result of this is that the patent life usually extends up to only 5 years before the generic competition starts. Ultimately, this results in pharmaceutical companies setting increasingly higher tariffs to make their new drugs sufficiently profitable to satisfy shareholders.
According to the Wellcome Trust (a global charitable foundation based in the United Kingdom), the drug development timeline can be outlined as: discovery, preclinical, clinical trials, approval, and post-launch.
We will describe the first two, the drug discovery process and preclinical development of a drug, within the context of cancer research. These comprise a number of stages that can be summarized as follows:
- Target identification and validation;
- Lead identification: in vitro screening (toward the target and cancer cell lines) of large numbers of chemical entities (potential drugs) to find hits (modulators of the target and cell growth inhibitors) which result in lead identification. Filing a patent is likely carried out at this stage;
- Lead optimization: in vitro screening; PK, PD, and ADMET studies; in vivo efficacy; and acute toxicity – initial evaluation;
- Development of candidate and progression to clinic: bioavailability, duration of action, laboratory scale synthesis, toxicity, and safety.
The differences in the time spent on the different steps in different medicinal chemistry projects are dictated by the relative resources available for each part of the process and the strategies employed by the researchers involved.
We describe these different steps and address them with several examples of anticancer drugs that have reached at least trial stages, and the history behind their discovery. Most of the names will be familiar, like cisplatin, taxol, and rituximab.
Selecting the Target
There appears to be an established premise in drug research nowadays: no medicinal chemistry project readily attracts interest from either for- or not-for-profit agencies prior to target identification and – increasingly – validation (in cancer models). Put simply – first find and justify your target. Cancer targets in the present context are those identified biomolecules involved in the numerous cellular processes of carcinogenesis.
The medicinal chemist does not identify (i.e. discover and validate) molecular targets, although target discovery has become a conventional part of the drug discovery process. The choice of the target then governs the direction of the chemistry of the project. Knowledge of the existence and function of a target is generally provided by physicians, pharmacologists, geneticists, and molecular and chemical biologists. However, once a clear target has been identified in one or more of the multiple steps of carcinogenesis, it is compelling for the scientific community to try to find a drug or modulator for it, and indeed this can trigger a discovery program. In fact, despite the need for validation of the target, knowing that a gene is mutated and selected for in cancer cells might be persuasive evidence to initiate a drug discovery project.
The selection of the target is not trivial. From a medicinal chemist’s point of view, the nature of the target is crucial for drug design, since this will affect the physical and chemical interactions of the pharmacophore (described by Paul Ehrlich in 1909 as the “molecular framework that carries the essential features responsible for a drug’s biological activity”) with the active site of the biomolecule. Additionally, the target is within a biological system, and reaching it with the “intact” drug or a derivatized drug (a pro-drug) is a major challenge for the medicinal chemist. For example, some targets will be accessible to the drug from the extracellular environment, but, on occasion, the target will be intracellular, and the drug must penetrate cellular membranes.
Since cancers are heterogeneous, involving different tissues of origin and differing molecular routes taken to achieve malignancy, the number of potential targets is considerable. Each chapter in this book contains a plethora of potential cancer drug targets. Because of the rapid progress made in identifying specific molecular targets in cancer and because of the central importance of this area to cancer biology, we have devoted a large part of Chapter 16 to a discussion of targeted agents and the new concepts that have arisen around their discovery and clinical use.
Rituximab (Rituxan), one of the most successful bench-to-bedside stories, was the first monoclonal antibody to gain FDA approval (1997) for the treatment of cancer, and the first single agent approved for lymphoma-targeted therapy (CD20-positive, β-cell, low-grade, or follicular non-Hodgkin lymphoma). Rituximab is a chimeric mouse–human antibody that recognizes and binds to cells expressing the CD20 antigen on the surface of malignant and normal B cells. Patients who do not respond well to rituximab treatment can be treated with antibodies labeled with radionuclides, additionally damaging cancer cells (90Y-ibritumomab, tiuxetan tositumomab, and 131I tositumomab).
Imatinib (Gleevec) is a small molecule and arguably the most successful drug in the young history of targeted therapies. A somatically mutated gene, BCR-ABL, is causally involved (through its product protein) in the carcinogenesis of a particular malignancy (chronic myeloid leukemia, CML). The inhibition of the aberrant protein BCR-ABL results directly in disease control, giving the first example of a success story of a targeted therapy. The discovery of imatinib is described in Box 16.1. Development of Imatinib is considered by some as the dawn of the molecular era of targeted therapy directed against oncogenic mutations. Second-generation BCR-ABL inhibitors include nilotinib, dasatinib, and bosutinib.
Other therapies based on this model include semaxanib (targeting VEGFR signaling), sorafenib, sunitinib, pazopanib, axitinib (targeting VEGFR), gefitinib, erlotinib (EGFR antagonists), flavopiridol (the first cyclin-dependent kinase inhibitor (CDKI) in trials), R-roscovitine (also a CDKI), bortzomib (proteasome inhibitor), cilengitide, Nutlin-3, and PRIMA-1. Some of these are discussed in more detail in Chapter 16.
Strategies to Find Lead Compounds
Once the target has been chosen, the first phase of the drug discovery process is the so-called hit-to-lead phase. The issue was neatly summarized by the medicinal chemist Frank King, who wrote in 2002:
It can be argued that the most important decision in drug discovery is that which is made every day by medicinal chemists; what compound to make next. Strategically, the two extremes to lead optimization and hence decision making are: rational design (careful design of single molecules using target structural information, pharmacophore identification and SAR [structure–activity relationships]) and random synthesis (make all possible analogues from readily available starting materials and trust luck). In practice, we do a combination of both, although the relative proportion of rational:random increases as the knowledge of SAR develops.
In this phase, hundreds of thousands of compounds are often screened against the target via at least one carefully selected test. A return of around 0.1% for hits is usually considered a success for molecular-based screening programs. The test is ideally carried out using high-throughput screening (HTS) where the appropriate choice of test is crucial, as interpretation of data may be difficult. Furthermore, poor choice of the screening assay may lead to overly positive evaluation that can be a carryover problem in the project.
In 1975, the NCI adopted the mouse P388 (leukemia) model as a primary screen. The model provided an indication of anti-tumor activity in a living system, yet because it represented only one tumor type, it was not always a good predictor of activity, did not guarantee reproducible success in humans, and, most importantly, could not be used in HTS. Human xenografts (human tumors growing in immunocompromised animals) presented similar problems. The need for an in vitro system that could provide researchers with solutions to these problems was addressed by the development of human tumor cell line screening. It was launched by the NCI in 1985, and used as primary routine screening since 1990. The screen includes 60 human tumor cell lines, representing leukemia, melanoma, and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney. The panel provides the identification of compounds with activity against human cancers of a particular tissue. Most importantly, these systems allow for HTS, affording both time and economic value. Unsurprisingly, there are some drawbacks as laboratory-grown cells might undergo adaptive changes, raising concerns as to whether they are truly representative of the cancer phenotype. In addition, cell-culture systems are limited in their ability to mimic many aspects of the in situ tumor microenvironment, including hypoxia, stromal cell interaction, and vascularization (Chapters 12 and 14). Nevertheless, cell culture systems have been extremely valuable tools for the discovery and evaluation of potential hits and have fostered the development of enhanced analytical tools such as the COMPARE algorithm developed by Kenneth Paull and colleagues at the NCI. Data from in vitro cytotoxicity screening of large numbers of compounds provide easy-to-interpret dose–response curves. Sensitive cancer cell lines are readily identified, and more importantly it has been proven that two agents with similar selectivity patterns toward the panel are most likely to have a similar mechanism of action.
Screening compounds against selected molecular targets is a complementary technique in which interaction (inhibitory potency and selectivity) of the target with a potential hit is examined. The identification of compounds that hit the target is followed by retests and generation of dose–response curves which provide chemists with IC50 values for those hits. At this point quantitative assessment of binding affinities is very important, including stoichiometry of binding, conformational changes, and identification of promiscuous inhibitors. This can lead to an investigation of more hits and larger libraries. Usually, a different assay is carried out on confirmed hits in a cellular environment.
The next step following identification of a series of hits is the generation or determination of the lead or leads of the series. Analogues are synthesized (or purchased where possible), and a quantitative structure–activity relationship (QSAR) is determined. The lead must have improved potency, reduced off-target activities, and physiochemical properties suggestive of reasonable in vivo PK. The lead is determined through empirical modification of the hit structure.
But what is the origin of the hits, and where do the original compounds from which the hits are selected come from? In fact, many different strategies are used to identify leads. Our classification of these strategies is based on the class of compounds from which they were first screened in the search for hits.
The source of hits, and hence the lead, may be: natural products (less productive but still highly valuable in oncology), established drugs (for “me-betters,” valid but commercially risky since this approach relies upon finding a novel, differentiated series of compounds, the benefits of which must translate into the clinic), pharmacological tools (HTS gives most of the leads), competitor patents, publications, or even serendipity. We have also included in this text a “Miscellaneous” section, where we describe light-activated therapeutics, RNA interference (RNAi), and vaccine therapies, and go on to explore the periodic table.
Serendipity
Although a serendipitous discovery of a hit series can hardly be classified as one of the intended strategies to find anticancer drugs, the importance of serendipity over the last 60 years in the discovery of drugs such as cisplatin and tamoxifen is undeniable.
Cisplatin
The discovery of cisplatin as an anticancer drug serendipitously arose from research on the effect of electric fields (generated with Pt electrodes) on the growth of bacteria in Barnett Rosenberg’s laboratory at Michigan State University in the early 1960s. Dr Rosenberg was a biophysicist investigating whether the telophase stage of mitosis, which seemed to have some visual similarity with the lines of force between the poles of a magnet, might have a magnetic component. He speculated that cell division may be affected by the magnetic field created by an electric current.
There was indeed an effect on the growth of Escherischia coli (E. coli) bacteria. The application of an electric current to the culture made the bacteria grow in a spaghetti-like fashion; however, they did not divide. Replication was halted. The results were reproduced in a series of experiments in which various strengths of current were applied. What was inhibiting cell replication? Was it the current? The researchers discovered that the bacterial filamentous growth was not a result of the electric field but of a chemical formed in the cell culture medium due to electrolysis of the platinum electrodes in the presence of components of the medium (Cl− and NH3). This gave rise to cis-diamminetetrachloridoplatinum(IV), a pro-drug for what we now know as cisplatin (Fig. 15.5a). In 1968, the first tests of cisplatin in mice bearing Sarcoma 180 solid tumors were carried out. Instead of treating the mice on the day after the tumor was implanted (standard protocol at the time), Rosenberg and his assistant Van Camp waited for 7 days, until the tumor had grown to about 1 g in weight (a 20-fold weight increase). The results were exceptional, producing a high percentage of complete cures. Rosenberg presented the results to the NCI, who verified the potent anti-tumor activity of cisplatin. The drug progressed from activity in mouse models in 1968 to human trials in 1971 and FDA approval in 1978.
Figure 15.5 (a) Structure of clinically used drug cisplatin. (b) Guanine nucleobase. (c) Intrastrand cis-{Pt(NH3)2(GG)} crosslink in a head-to-head conformation as part of the adduct formed from the reaction of cisplatin with the duplex 5′-d(TCTCGGTCTC)·d(GAGACCGAGA). Platinum is light blue, nitrogens coordinating to platinum are deep blue, oxygens C6O in the crosslinked guanines are red, and other atoms are gray for clarity.
(Coordinates taken from the Protein Data Bank, 1AIO.)
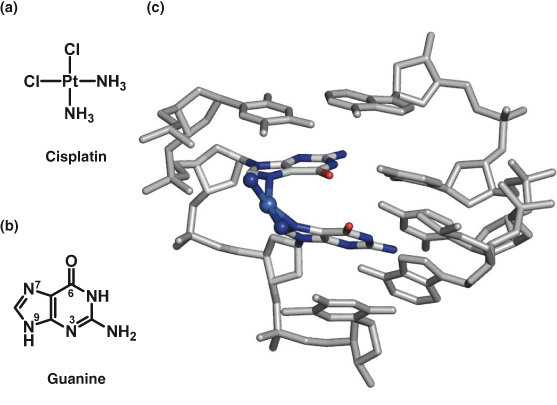
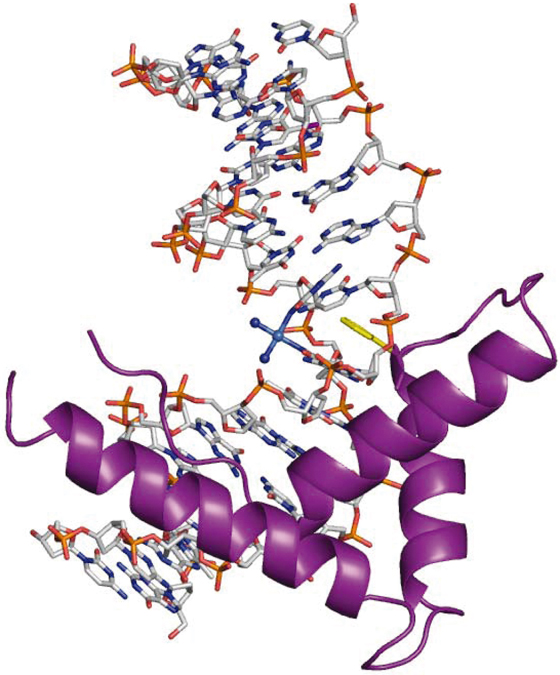
Although the discovery of the anticancer properties of cisplatin was serendipitous, we now have a wealth of information regarding the cellular processing and mechanism of action of the drug. DNA is accepted as its intracellular target, but it also binds to RNA and proteins. In its interaction with DNA, cisplatin targets guanine-rich sequences since it binds strongly to N7 in the guanine nucleobase (the most electron-dense and accessible site on DNA for electrophilic attack by platinum; Fig. 15.5b). Both chlorido ligands bound to platinum in cisplatin can be substituted by water molecules (hydrolyzed) in aqueous environments and these weakly bound water molecules can be further substituted by more strongly binding ligands such as guanines (Fig. 15.5c). Cisplatin readily forms bifunctional intrastrand GG crosslinks on DNA, believed to be lethal lesions. These intrastrand crosslinks cause DNA to bend. Such bent DNA is recognized by intracellular (HMG) proteins which can play a role in protecting the DNA from repair (Fig. 15.6). Additionally, this DNA perturbation may hijack proteins that recognize DNA kinks from their natural locus and contribute to the anti-tumor effects of cisplatin. Ultimately, the DNA damage results in apoptosis and cell death.
Figure 15.6 Adduct formed between the non-sequence-specific domain A of HMG1 and cisplatin-modified DNA. Platinum is light blue, nitrogen is deep blue, oxygen is red, phosphorus is orange, carbon is gray, and HMG is a purple ribbon, with a phenylalanine intercalating between two nucleobases highlighted in yellow.
(Coordinates taken from the Protein Data Bank, 1CKT.)
The story of John Cleland, patient zero for what would become the standard cisplatin treatment for men with advanced testicular cancer, was recorded in an interview for the magazine Cure Today in 2004 (winter issue), 30 years after he began the cisplatin treatment that cured him.
Tamoxifen
Tamoxifen (Nolvadex-D, now generic; Fig. 15.7) is an early exemplar of a targeted drug. It is a selective estrogen-receptor modulator (SERM), discovered during investigations of the role of estrogen in breast cancer and as a result of intense research in the 1950s to find a contraceptive. It evolved to become the first targeted therapy for the treatment of breast cancer and is considered by some as the most established case of tumor-tailored therapy. However, it had a rather tortuous journey to its pole position in cancer therapy.
Tamoxifen was first synthesized by Richardson in 1962 in the Alderley Park research laboratories of ICI Pharmaceuticals (now AstraZeneca). Walpole, who led the team in reproductive endocrinology where Richardson was working and who was trying to find a contraceptive, filed a UK patent primarily covering an invention for application on “the management of the sexual cycle.” Although cancer treatments were not a corporate priority at the time, Walpole’s personal interest in cancer therapies inspired him to include in the patent coverage an application for the “control of hormone-dependent tumours.” After a rather quiet evolution, tamoxifen reached a critical point in its development in 1972, when ICI Pharmaceuticals almost terminated tamoxifen due to negative prospects for market exploitation. The crisis point was overcome by Walpole’s tenacity, and ICI Pharmaceuticals marketed tamoxifen in the United Kingdom as a breast cancer treatment in 1973 and as an inducer of ovulation in 1975. The US patent for the treatment of advanced breast cancer in postmenopausal women was filed in 1977.
Certainly the 1970s were quite a dark era for tamoxifen. It enjoyed little interest from either medical advisors or the pharmaceutical industry right up to the mid-1980s. However, during those dark times, Jensen discovered the estrogen receptor and Jordan developed the application of the estrogen-receptor assay to predict endocrine responsiveness to endocrine ablation successfully. During those times, its mechanism of action was explained at the molecular level. It was discovered that tamoxifen, which has low affinity for the estrogen receptor, was in fact a pro-drug that accumulated and was then converted to 4-hydroxytamoxifen, the active metabolite with high affinity for the estrogen receptor and anti-estrogenic activity. A good example of how important patent protection becomes in drug development is the fact that the publication of these data in 1977 had been delayed for more than a year to secure patent protection for the metabolites, since tamoxifen did not have patent protection in the United States at the time.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


