Bone Tumors
Bogdan Czerniak
Tomasz Tuziak
Andrzej Kram
Alberto Ayala
Primary bone tumors, excluding lymphohematopoietic malignancies, constitute only 0.2% of all tumors and occur at a rate of approximately one tenth that of the closely related soft-tissue sarcomas. The overall incidence rate is approximately 0.8/100,000 (Dorfman and Czerniak, 1995). The first well-defined peak occurs during the second decade of life, and the second peak occurs in patients older than 60 years (Fig. 36-1). This bimodal age-specific incidence rate pattern of bone sarcomas is clearly different from that of soft-tissue sarcomas, which gradually increases with age.
Because bone tumors of different types often have overlapping microscopic features, evaluating them without clinical and radiographic context may result in an incorrect diagnosis. Therefore, they should be approached as clinicopathological entities whose behavior and biological potential must be assessed by both microscopic details and clinical factors such as patient age, location in a particular skeletal site, specific radiographic presentation, and association with underlying conditions (Sanerkin and Gallagher, 1979). As is the case with soft-tissue sarcomas, we advocate a multimodal approach to establish the diagnosis with a stepwise analysis of clinical, radiographic, microscopic, and, in selected cases, molecular data. Such a multidimensional analysis is more likely to result in a clinically valid assessment of the lesion (Saeter, 2003).
A diagnostic specimen can be obtained by open biopsy or various transcutaneous (closed) biopsy techniques (de Santos et al, 1978). The use of the latter techniques, which are often assisted by radiographic imaging techniques, has dramatically increased in many institutions. Aspiration cytology, combined with core-needle biopsy, is often used as a preliminary diagnostic approach to bone lesions, and in many cases it yields adequate material to establish the final diagnosis (deSantos et al, 1979b; Ayala et al, 1995; Collins et al, 1998a; Agarwal et al, 2000). However, some bone lesions are extremely difficult or even impossible to diagnose on the basis of the small amount of material obtained by core-needle biopsies or fine-needle aspirates. In such instances, an open biopsy must be performed.
Aspiration biopsy of bone lesions is one of the oldest diagnostic applications of this technique. Coley et al (1931) described their findings in 35 patients, with remarkably accurate results. In Argentina, Schajowicz and Lemos (1970) collected several thousand patients for whom the primary diagnosis was established by aspiration rather than by open biopsy. Because the cytologic sampling may be tedious, the
contemporary trend is to perform core-needle biopsies under radiologic guidance and examine the residual material in the form of smears.
contemporary trend is to perform core-needle biopsies under radiologic guidance and examine the residual material in the form of smears.
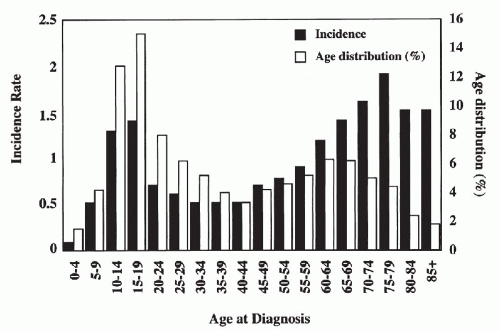 Figure 36-1 Bone sarcomas: epidemiology. Age-specific incidence rate and frequency distribution, all races, sexes, SEER data, 1973-1987. |
Except for bone lesions in multiple myeloma, which are routinely aspirated for diagnosis, the aspiration biopsy is principally used to confirm the nature of suspected, relatively uncommon, primary, or (more commonly) metastatic bone tumors. In metastatic cancer, the site of origin may be sometimes determined in smears. Aspirations are rarely used to diagnose inflammatory or reactive conditions of the skeleton, which include osteomyelitis (caused by Staphylococcus or Mycobacterium tuberculosis), exuberant fracture callus, and other trauma-related lesions.
Planning the biopsy approach (open or closed) is a complex process that must take into account the technical aspects of subsequent definitive surgery. An inappropriately selected biopsy site may preclude limb-sparing definitive surgery. In general, the location of the biopsy track should be such that if the lesion proves to be malignant, it can be excised en bloc with the segment of affected bone. This is particularly important if limb-salvaging procedures are to be successful. It is generally recommended that preoperative diagnostic procedures should be performed at medical institutions that can provide definitive treatment (Simon et al, 1986; Raymond et al, 1995).
In this chapter, we provide an overview of bone lesions, focusing on those conditions that are primary targets for aspiration cytology. Benign lesions will be discussed only as points of differential diagnosis.
TECHNIQUES OF ASPIRATION BIOPSY
Aspiration biopsy of bone lesions has special requirements. To perforate the cortical bone, a thick needle (external diameter = 1.2 to 1.5 mm) with a stylet is used as a guide for the thin needle. The skin and the subcutaneous tissue are locally anesthetized to the level of the periosteum. Subsequently, the thick needle is inserted through the bone to the periphery of the lesion. The stylet is withdrawn and a thin needle is introduced into the target through the guide. The aspiration then proceeds as in any other organ (see Chap. 28).
A brief period of general anesthesia is required under the following circumstances:
The tumor causes a great deal of pain or is painful on palpation.
A vertebral body is the target of aspiration.
The aspiration is performed on a child.
Smaller lesions are often aspirated under the guidance of fluoroscopy or computed tomography (CT), which requires close cooperation with the radiologist. Large, palpable lesions of the long bones or the sternum can be aspirated without x-ray monitoring.
Thin-needle aspirates of bone lesions often yield a large amount of blood that may enter the lumen of the syringe. Smears prepared from such material are commonly of scant cellularity. The methods used to secure diagnostic cells from bloody specimens are described in Chapter 28.
OSTEOBLASTIC LESIONS
Osteoblastic or bone-forming lesions can be divided into three major categories. The first category includes lesions that are clinically benign and practically never recur after simple excision. The intermediate group consists of lesions with locally destructive growth and a high recurrence rate,
but virtually no metastatic potential. The third category comprises frankly malignant tumors with a high propensity for distant metastasis. Some locally aggressive bone-forming tumors may progress, typically after many recurrences, to highly aggressive sarcomas. The phenomenon is similar to the progression of low-grade chondrosarcoma to dedifferentiated chondrosarcoma (Abdelwahab et al, 1997; Reith et al, 1999). Among bone-forming lesions, areas of anaplasia are frequently seen in low-grade intramedullary osteosarcoma and low-grade bone surface (parosteal) osteosarcoma (Wold et al, 1984; van Oven et al, 1989; Abdelwahab et al, 1997; Shuhaibar and Friedman, 1998; Reith et al, 1999).
but virtually no metastatic potential. The third category comprises frankly malignant tumors with a high propensity for distant metastasis. Some locally aggressive bone-forming tumors may progress, typically after many recurrences, to highly aggressive sarcomas. The phenomenon is similar to the progression of low-grade chondrosarcoma to dedifferentiated chondrosarcoma (Abdelwahab et al, 1997; Reith et al, 1999). Among bone-forming lesions, areas of anaplasia are frequently seen in low-grade intramedullary osteosarcoma and low-grade bone surface (parosteal) osteosarcoma (Wold et al, 1984; van Oven et al, 1989; Abdelwahab et al, 1997; Shuhaibar and Friedman, 1998; Reith et al, 1999).
Benign osteoblastic lesions were first recognized as a distinct group by Jaffe and Mayer (1932). The identification of osteoid osteoma as a distinct entity came later, in a report by Jaffe (1935). Jaffe (1956) and Lichtenstein (1956) independently described osteoblastoma to delineate osteoblastic tumors with greater growth potential than osteoid osteoma. More recent experience with benign osteoblastic tumors has indicated that some lesions may reach a considerable size, exceeding 4 cm in diameter. These lesions have a locally destructive growth pattern with a high recurrence rate after curettage, and are referred to as aggressive osteoblastomas (Miyayama et al, 1993).
The three main categories of benign osteoblastic tumors—osteoid osteoma, osteoblastoma, and aggressive osteoblastoma—represent a continuum of lesions with different growth potentials, levels of extrinsic humoral activity, and recurrence rates (Fig. 36-2). Osteoid osteomas and osteoblastomas have nearly identical microscopic features and thus cannot be distinguished from one another by microscopy alone. Some authors have proposed that lesions larger than 1.5 cm in diameter should be classified as osteoblastomas. In contrast, aggressive osteoblastoma, in addition to its larger size (>4 cm), is characterized by the presence of so-called epithelioid osteoblasts (Schajowicz and Lemos, 1970; Lucas et al, 1994).
Osteosarcoma is a prototypic malignant bone-forming tumor, and the most common primary sarcoma of bone. This term is used to describe a heterogeneous group of lesions with diverse morphologies and clinical behaviors. Designations such as osteoblastic, chondroblastic, and fibroblastic osteosarcoma reflect the microscopic variability of the tumors and the presence of histologically different components within one lesion (Inwards and Unni, 1995). Osteosarcoma may originate and grow primarily inside the bone (intramedullary osteosarcoma) or it may grow on the surface on bone (surface osteosarcoma) within periosteal or paraosteal tissue (Ahuja et al, 1977; Bertoni et al, 1982; Okada et al, 1994). The currently recognized types of osteosarcoma and the descriptive terms used in their diagnosis are listed in Table 36-1.
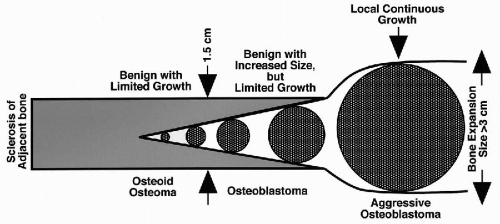 Figure 36-2 Benign osteoblastic tumors. Comparative features of biologic behavior and growth. (From H. Dorfman and B. Czerniak, Bone Tumors, Mosby, Inc., St. Louis, 1998). |
Osteoid Osteoma
Osteoid osteoma is a benign tumor that consists of a well differentiated bone-forming nidus surrounded by a distinct zone of reactive bone sclerosis. The nidus has limited growth potential and usually is less than 1 cm in diameter (Jaffe, 1935; Gitelis and Schajowicz, 1989). Osteoid osteomas are relatively common lesions that account for approximately 10% of all primary bone tumors. They usually affect teenagers and young adults at a male-to-female ratio of 3:1. Osteoid osteoma has characteristic, often diagnostic, clinical symptoms. In the vast majority of cases, patients present with pain of increasing severity that is relieved by aspirin or other nonsteroidal antiinflammatory agents. Approximately 50% of osteoid osteomas involve the long bones of the lower extremity, with the femoral neck being the most frequent site (Gitelis and Schajowicz, 1989.) They also frequently affect the small bones of the hands and feet. In contrast to osteoblastomas, they occur infrequently in the spine. Microscopically, the nidus consists of an interlacing network of bone trabecule with different levels of mineralization rimmed by prominent osteoblasts with occasional multinucleated giant cells (Schajowicz and Lemos, 1970).
Osteoblastoma
Osteoblastomas occur approximately one third less frequently than osteoid osteomas. They affect individuals with an age peak incidence in the second decade of life, and the male-to-female ratio is approximately 3:1. These tumors have a predilection for the axial skeleton and frequently involve the vertebral column, sacrum, and craniofacial bones (Schajowicz and Lemos, 1970).
Radiographically, osteoblastomas are similar to osteoid
osteomas. They present as a lytic, well demarcated lesion surrounded by a zone of reactive sclerosis with a nidus exceeding 1.5 cm in diameter (Jaffe, 1956; Lichtenstein, 1956) (Fig. 36-3A).
osteomas. They present as a lytic, well demarcated lesion surrounded by a zone of reactive sclerosis with a nidus exceeding 1.5 cm in diameter (Jaffe, 1956; Lichtenstein, 1956) (Fig. 36-3A).
TABLE 36-1 TYPES OF OSTEOSARCOMA AND DESCRIPTIVE TERMS USED IN THEIR DIAGNOSIS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Grossly, the nidus appears as a granular, friable, reddish tissue with secondary changes due to hemorrhage and cystic degeneration.
Microscopically, the nidus is similar to an osteoid osteoma nidus, consisting of a network of bone trabeculae distributed in loose fibroblastic stroma with prominent vasculature (Lucas et al, 1994) (Fig. 36-3B). Multinucleated giant cells and rimming osteoblasts are present (Schajowicz and Lemos, 1970; Ruggieri et al, 1996).
Aggressive Osteoblastoma
Pathology and Histology
Aggressive osteoblastomas are extremely rare tumors that are considered as borderline lesions between benign osteoblastomas and osteosarcomas (Dorfman and Weiss, 1984). They have a higher growth potential than conventional osteoblastomas, and typically exceed 4 cm in diameter. Clinically, aggressive osteoblastomas are characterized by destructive growth and high risk for recurrence, but they have virtually no metastatic potential.
Microscopically, the presence of so-called epithelioid osteoblasts is the main histological difference between conventional and aggressive osteoblastomas (Miyayama et al, 1993). Epithelioid osteoblasts are round cells with abundant eosinophilic cytoplasm and an eccentrically located, large, oval nucleus with a prominent nucleolus. The nucleus is often displaced by a clear cytoplasmic area, which ultrastructurally represents an enlarged Golgi apparatus.
Cytology
Osteoid osteomas are practically never diagnosed by cytology. Aspirations of benign osteoblastic lesions are typically performed on larger lesions involving deep anatomic sites that require major surgery for an open biopsy (Fig. 36-3A). Aspirations from osteoblastoma are highly cellular and contain oval osteoblastic cells with eccentric nuclei and dense eosinophilic cytoplasm (epithelioid osteoblasts) (Fig. 36-3D). Some of these cells contain a well demarcated cytoplasmic halo representing a prominent Golgi center, a feature frequently seen in activated osteoblastic cells (Fig. 36-3, upper inset). Scattered multinucleated giant cells are usually present. Mitotic figures are rare. No atypical mitoses or nuclear pleomorphism can be seen. Aggressive osteoblastoma should be considered if an aspirate from an osteoblastic lesion, exceeding 4 cm in diameter, contains a large population of enlarged epithelioid osteoblasts with some nuclear pleomorphism and more than occasional mitotic figures.
Osteosarcoma
Pathology and Histology
Osteosarcoma is a malignant tumor of bone in which malignant mesenchymal cells have the ability to produce osteoid matrix or immature bone (Unni, 1998).
It accounts for approximately 20% of all primary sarcomas of bone, excluding multiple myeloma and hematopoietic neoplasms. The most frequent sites of skeletal involvement and the peak age incidences are shown in Figure 36-4. The age distribution is bimodal, with the first peak occurring during the second decade of life, and the second, smaller peak occurring in patients older than 50 years. The incidence within the specific bone corresponds to the site of greatest growth rate. Accordingly, the distal femoral and proximal tibial metaphyses are the most common sites for osteosarcoma. The humerus is the third most frequently involved bone, with the majority of cases involving the proximal humeral metaphyses. Other, less frequent locations include the axial skeleton, craniofacial bones, and pelvis (Clark et al, 1983). This description is limited to the most frequent conventional high-grade intramedullary form of osteosarcoma frequently diagnosed by aspiration cytology.
A more comprehensive description of all variants of osteosarcoma is beyond the scope of this review.
A more comprehensive description of all variants of osteosarcoma is beyond the scope of this review.
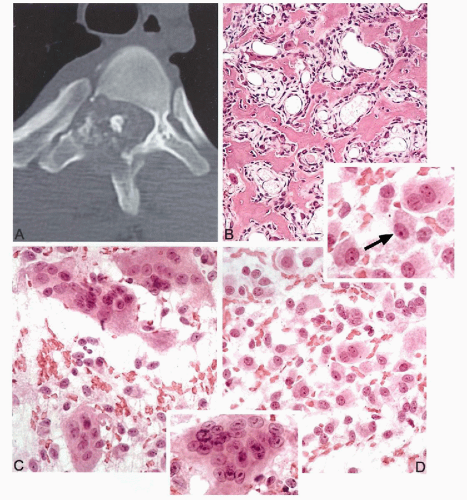 Figure 36-3 Osteoblastoma of the vertebral column. A. Computer tomogram shows a lytic destructive tumor containing foci of mineralized bone matrix that involve the pedicle and lamina of the thoracic vertebra. It expands into the spinal canal, displacing a dural sac and compressing the spinal cord. B. Microscopic features of osteoblastoma composed of irregular bony trabeculae rimmed by prominent osteoblastic cells. Note the stroma with prominent dilated vascular channels and scattered multinucleated osteoclastic giant cells. C. Fine-needle aspirate showing scattered multinucleated giant cells and multiple mononuclear epithelioid osteoblastic cells with a prominent eosinophilic cytoplasm. Inset. Higher magnification of a multinucleated giant cell with centrally clustered oval nuclei containing prominent nucleoli. D. High magnification of epithelioid osteoblastic cells with prominent oval densely eosinophilic cytoplasm and peripherally located nuclei containing discrete nucleoli. Note that some of the cells contain double nucleoli. Inset. Higher magnification of osteoblasts. Some of them show ill-defined cytoplasmic lucency (arrow) corresponding to a prominent Golgi center. (B-D: H&E.) |
Although the vast majority of osteosarcomas are de novo lesions, approximately 10% to 15% of them arise in patients with rare clinical syndromes, such as familial osteosarcoma, retinoblastoma-associated osteosarcoma, Rothmund-Thomson syndrome, and multifocal osteosarcoma. Some tumors may develop in association with several neoplastic and nonneoplastic precursor lesions (Dick et al, 1982; Haibach et al, 1985; Hansen et al, 1985). Familial osteosarcoma occurs in several generations of families that are most frequently affected by retinoblastoma (Draper et al, 1986). Li-Fraumeni syndrome is another familial cancerpredisposing syndrome in which a germ line mutation of
the tumor-suppressor gene p53 is associated with a high incidence of osteosarcoma (Garber et al, 1991). Rothmund-Thomson syndrome is a very rare condition characterized by lesions of the skin, eye, genitals, central nervous system, and bone. This disorder predisposes an individual to squamous cell carcinoma of the skin and osteosarcoma, which may be multicentric.
the tumor-suppressor gene p53 is associated with a high incidence of osteosarcoma (Garber et al, 1991). Rothmund-Thomson syndrome is a very rare condition characterized by lesions of the skin, eye, genitals, central nervous system, and bone. This disorder predisposes an individual to squamous cell carcinoma of the skin and osteosarcoma, which may be multicentric.
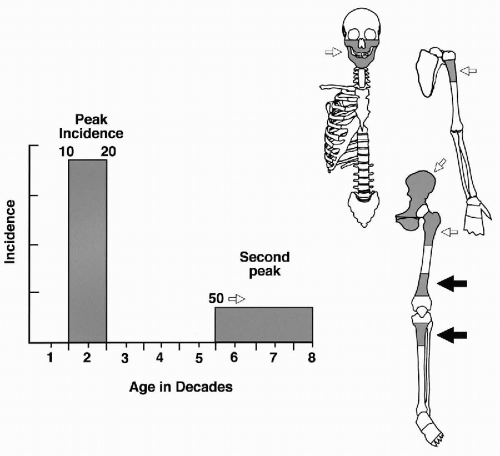 Figure 36-4 Osteosarcoma: peak age incidence and frequent sites of skeletal involvement. The most frequent sites are indicated by solid black arrows. (From H. Dorfman and B. Czerniak, Bone Tumors, Mosby, Inc., St. Louis, 1998.) |
Paget’s disease is the most frequent nonneoplastic precursor lesion of osteosarcoma in older patients (Haibach et al, 1985). Osteosarcomas complicating Paget’s disease are usually of high grade and most frequently involve the pelvis, humerus, and femur. In general, secondary osteosarcomas that complicate predisposing conditions are associated with a significantly worse prognosis then conventional de novo high-grade osteosarcomas. Radiation-induced osteosarcomas develop with a latency period of 3 years or more after radiation treatment; most such tumors are of high grade and follow an aggressive course with short survival (Sim et al, 1972). Bone infarcts rarely give rise to osteosarcomas. A small number of high-grade osteosarcomas have been reported in patients who have received metallic prosthetic implants (Martin et al, 1988). Other nonneoplastic conditions, such as fibrous dysplasia and chronic osteomyelitis, are associated with a very low incidence of secondary malignancy (Johnston and Miles, 1973).
Current multidrug chemotherapeutic regimens substantially reduce tumor mass in at least 50% of patients; in some of these patients, complete or near complete necrosis can be accomplished. The latter is associated with a substantially better prognosis compared to a tumor that responds less favorably to chemotherapy (Raymond et al, 1987). Thus, pathologic assessment of chemotherapy effect is now universally accepted as an integral element of the multimodal approach (Ayala et al, 1984). Moreover, the degree of necrosis in the postchemotherapy specimen is used as a factor in modifying the postoperative treatment protocol (Raymond et al, 1995).
The radiographic presentation of osteosarcoma varies greatly, from completely lytic to sclerotic lesions, but typically the combination of these features enables a radiographic diagnosis to be established (Sweet et al, 1981; deSantos and Edeiken, 1982; Sundaram et al, 1987). The production and mineralization of the bone matrix by the tumor results in cloudy opacities that vary in size, shape, and density (Fig. 36-5A). These opacities may be uniformly distributed throughout the lesion or they may cluster in one or several areas of the tumor. Occasionally, osteosarcoma can present as a large, solid, ivory-like sclerotic mass with heavy mineralization. The growth pattern is destructive, with ill-defined boundaries between the tumor and normal bone (Madewell et al, 1981). Usually the cortex is at least partially, or more often completely, disrupted. In such instances, the tumor extends to subperiosteal tissue and elevates the periosteum, forming the so-called Codman’s triangle. An obvious extension of tumor into the periosteal soft tissue is seen in the majority of such cases (deSantos et al, 1979a; Ragsdale et al, 1981).
The gross appearance of osteosarcoma is best described in its typical location, i.e., in the metaphyseal portion of a long bone. A combination of bony and soft-tissue areas is responsible for the heterogeneous appearance of the tumor, which has areas that vary in color, consistency, and degree of ossification (Fig. 36-5B). Heavily ossified areas are ivorycolored and can be as hard as normal cortical bone. Less ossified lesions are soft, fleshy, and lighter yellow (deSantos and Edeiken, 1982). The tumor exhibits an invasive, bone-destructive growth pattern. Small lesions are usually eccentrically located and attached to the inner surface of the cortex. More advanced lesions fill the medullary cavity and show evidence of cortical destruction with elevation of periosteum and extension into the soft tissue.
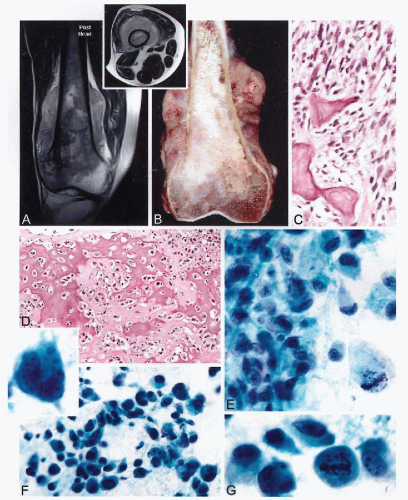 Figure 36-5 Osteosarcoma of the proximal tibial metaphysis. A. T1-weighted magnetic resonance (MR) image shows extensive cortical destruction and extension of the tumor into soft tissue and the epiphysis, with inhomogeneous signal. Inset. Coronal T1-weighted MR image shows an inhomogeneous medullary tumor with circumferential extension to the periosteal soft tissue. B. Gross photograph of frontal section of same tibial resection specimen after limb salvage procedure shows heavily ossified tumor with circumferential extension into the periosteal soft tissue. The tumor is heterogeneous with heavily ossified ivory-like areas centrally and less ossified soft-fleshy and gritty appearance peripherally. C. Histologic features of high-grade osteosarcoma showing spindle malignant cells producing immature osteoid. D. Histologic features of a high-grade osteosarcoma with abundance of immature tumor bone deposited by osteoblastic tumor cells. E. Fine-needle aspirate containing large irregular clusters of highly pleomorphic sarcomatoid cells. Inset. Abnormal mitotic figure. F. High magnification of anaplastic sarcomatoid cells forming loosely arranged clusters with brisk mitotic activity. Inset shows a multinucleated giant tumor cell. G. Higher magnification of anaplastic, somewhat epithelioid sarcomatoid cells with pronounced atypia and mitotic activity. (C-G: H&E.) |
The microscopic findings may vary considerably from lesion to lesion and from area to area. Evidence of direct production of an osteoid matrix by sarcomatous cells is required before a lesion can be classified as an osteosarcoma. The relations between two tumor components—the tumor cells and an extracellular matrix—are very important for diagnosis. According to the predominant type of matrix involved, the osteosarcoma is subdivided into three main categories: osteoblastic, chondroblastic, and fibroblastic (Dahlin and Unni, 1977; Unni and Dahlin, 1989). Osteosarcoma frequently presents as an undifferentiated sarcoma with predominant pleomorphic, round, or spindle cells showing only scattered foci of osteoid production. The tumor cells may have densely eosinophilic cytoplasm with eccentrically placed nuclei, and resemble osteoblasts, but are usually larger than normal or even activated osteoblasts. These cells may infiltrate the medullary cavity or extraosseous tissue in a dispersed manner or they may grow in large cohesive sheets with epithelioid features (Yoshida et al, 1989).
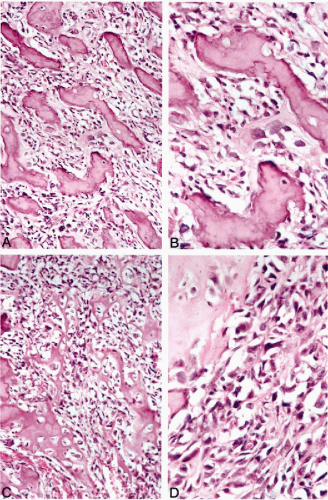 Figure 36-6 Patterns of osteoid depositions in osteosarcoma. A. Thick trabecula of osteoid between sarcomatoid tumor cells. B. Higher magnification of A showing coarse, well developed trabeculae of immature osteoid within sarcomatoid tumor cells. C. Early lace-like osteoid bands between sarcomatoid tumor cells. D. Higher magnification showing a border between anaplastic tumor cells and a solid area of osteoid. |
The osteoblastic nature of the tumor cells is best recognized by close appositions of sarcomatous cells to tumor bone trabeculae, or by their entrapment in lace-like osteoid depositions (Fig. 36-6A-D). It is important to remember that osteosarcoma can show the whole spectrum of sarcomatous
features, from small and undifferentiated, to oval and spindle-shaped, to highly pleomorphic neoplasms (Edeiken et al, 1987; Ayala et al, 1989; Nakajima et al, 1997). As in other high-grade sarcomas, a prominent vascular hemangiopericytoma-like pattern can be found. Numerous atypical mitoses are usually present. At the opposite end of the spectrum are highly ossified sclerosing lesions in which a full range of tumor bone production can be observed. Osteosarcoma can also exhibit cartilaginous differentiation. In some instances, the cartilaginous component may be extensive and dominant throughout the lesion, but even in predominantly chondroid tumors, at least focal direct osteoid production by sarcomatous tumor cells can be found. Tumors with predominantly chondroblastic features are frequently found in the craniofacial bones. The fibroblastic type of osteosarcoma is characterized by the presence of a predominant spindle-cell component similar to that seen in fibrosarcoma. The production of osteoid by fibroblast-like cells discloses the bone-forming nature of these lesions and helps to differentiate them from other spindle-cell neoplasms of the bone (see Table 36-1).
features, from small and undifferentiated, to oval and spindle-shaped, to highly pleomorphic neoplasms (Edeiken et al, 1987; Ayala et al, 1989; Nakajima et al, 1997). As in other high-grade sarcomas, a prominent vascular hemangiopericytoma-like pattern can be found. Numerous atypical mitoses are usually present. At the opposite end of the spectrum are highly ossified sclerosing lesions in which a full range of tumor bone production can be observed. Osteosarcoma can also exhibit cartilaginous differentiation. In some instances, the cartilaginous component may be extensive and dominant throughout the lesion, but even in predominantly chondroid tumors, at least focal direct osteoid production by sarcomatous tumor cells can be found. Tumors with predominantly chondroblastic features are frequently found in the craniofacial bones. The fibroblastic type of osteosarcoma is characterized by the presence of a predominant spindle-cell component similar to that seen in fibrosarcoma. The production of osteoid by fibroblast-like cells discloses the bone-forming nature of these lesions and helps to differentiate them from other spindle-cell neoplasms of the bone (see Table 36-1).
Cytology
Aspirates from osteosarcomas are usually, but not always, highly cellular and contain frankly malignant mesenchymal cells arranged in large clusters and smaller groups, or individually dispersed (see Fig. 36-5E-G). Osteosarcoma cells are spindled, oval, rounded, epithelioid, or pleomorphic, and show a high degree of cellular atypia. The malignant cells often have abundant dense cytoplasm and large nuclei containing prominent nucleoli that do not show any specific feature of osseous differentiation. The overall cytologic picture, in most instances, is indistinguishable from that of a pleomorphic malignant tumor, such as a malignant fibrous histiocytoma (Hakky et al, 1990). Cytologic preparations from osteosarcoma may contain variable amounts of collagen that mimics an osteoid. Fragments of cartilage matrix, as well as multinucleated giant cells, may also be present (Ellison et al, 1996). In general, a correlation of cytologic findings with clinical and radiographic presentations permits a preoperative cytologic diagnosis of osteosarcoma to be established in most cases (White et al, 1988).
TUMORS OF CARTILAGE
Tumors of cartilage can be benign or malignant. Benign cartilaginous lesions can be reactive, neoplastic, hamartomatous, or dysplastic in nature. In all of these conditions, proliferating cartilage cells may show cytologic atypia, and thus evaluations of these lesions that disregard the clinical and radiological contexts may lead to serious diagnostic errors (Bonnevialle et al, 1988). The rare cartilage-containing reactive lesions, such as synovial chondromatosis, florid reactive periosteitis, bizarre parosteal osteochondromatous proliferations, acquired osteochondroma, subungual exostosis, and pubic osteolysis, are all characterized by proliferation of cartilage, which may show pronounced atypia (Bendl, 1980; Spjut and Dorfman, 1981; Nora et al, 1983; Lindeque et al, 1990; Meneses et al, 1993; Edeiken et al, 1994). Chondroma is a prototypic, benign cartilage neoplasm that most frequently involves the medullary cavity and rarely presents as a subperiosteal (juxtacortical) lesion (Bauer et al, 1982). Enchondromatosis represents a dysplastic cartilage condition and occurs in two clinical settings: Ollier’s disease and Maffucci syndrome. The lesions of cartilage in these conditions may show pronounced atypia (Lewis and Ketcham, 1973; Cannon and Sweetnam, 1985). Chondroblastoma and chondromyxoid fibroma are two examples of benign neoplasms. They are characterized by the presence of immature cartilage cells, which may focally produce extracellular cartilaginous matrix. Developmental cartilage anomaly of the hamartomatous type is represented by osteochondroma, which may be solitary or multiple.
Chondrosarcoma is the term given to a heterogeneous group of cartilage tumors that are characterized by diverse morphologic features and clinical behaviors, which range from locally aggressive, slowly growing, nonmetastazing lesions to highly aggressive, lethal sarcomas (Sanerkin and Gallagher, 1979; Gitelis et al, 1981). Conventional chondrosarcoma is the most frequent malignant cartilage tumor, and it is further subdivided into three grades based on the level of nuclear atypia. Most chondrosarcomas are of the low and intermediate grades (grades 1 and 2). Since the degree of atypia in grade 1 chondrosarcoma may overlap with the atypia seen in benign chondroma, it is often impossible to differentiate these lesions without clinical and radiographic correlation. The special types of chondrosarcoma include dedifferentiated, clear-cell, and mesenchymal chondrosarcoma (McCarthy and Dorfman, 1982; Frassica et al, 1986; Laporte et al, 1996). These tumors have distinct morphologic features and clinical behaviors, and should be considered entities separate from conventional chondrosarcoma. The differences in the biologic potential of cartilage lesions, as related to their degree of differentiation, are provided in Figure 36-7.
Because of the presence of nuclear atypia in many benign neoplastic and metaplastic conditions that overlaps with atypia seen in malignant cartilage tumors, aspiration cytology has a limited application in the differential diagnosis of cartilage lesions. It is typically used as a preliminary diagnostic approach to radiographically identified lesions suspected to represent chondroblastoma, and, less frequently, chondromyxoid fibroma. It may also be used to rule out dedifferentiation in radiographically suspected high-grade chondrosarcoma. Reactive and metaplastic cartilage lesions are practically never diagnosed by cytology. Cytology is also not recommended as a diagnostic modality for the differential diagnosis of benign versus low to intermediate grade hyaline cartilage tumors, but it may be used to verify the nature of clinically and radiographically identified recurrent or metastatic lesions in patients with a history of treated chondrosarcoma.
Chondromyxoid Fibroma
Pathology and Histology
Chondromyxoid fibroma is composed of myxoid mesenchymal tissue, corresponding to early primitive phases of
cartilaginous differentiation. It accounts for less than 1% of all primary bone tumors (Feldman et al, 1970). Chondromyxoid fibroma has a predilection for metaphyseal parts of the long tubular bones, predominantly of the lower extremity.
cartilaginous differentiation. It accounts for less than 1% of all primary bone tumors (Feldman et al, 1970). Chondromyxoid fibroma has a predilection for metaphyseal parts of the long tubular bones, predominantly of the lower extremity.
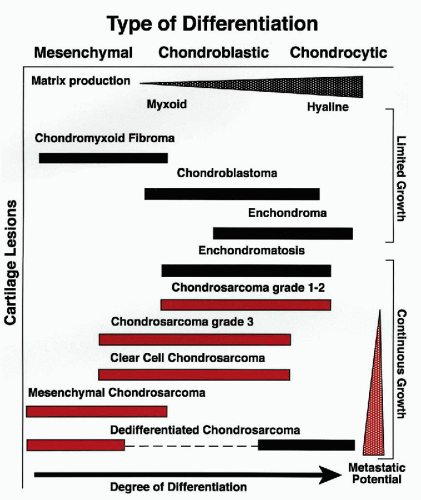 Figure 36-7 Differences in the biological potential of cartilage lesions. Graphic representation related to their degree of differentiation and clinical behavior. (From H. Dorfman and B. Czerniak, Bone Tumors, Mosby, Inc., St. Louis, 1998.) |
Approximately 30% of cases are diagnosed in the knee region, where the proximal tibial metaphysis is the most frequently involved site, followed by the distal femoral metaphysis. A unique subset of chondromyxoid fibromas involves the small bones of the acral skeleton, predominantly the feet. The pelvis, especially the ilium, is a frequently affected flat bone. Most patients are in their second or third decade of life, but there are scattered cases involving patients in the later decades of life (Batsakis and Raymond, 1989).
Radiographically, chondromyxoid fibromas are well demarcated eccentric lytic lesions whose long axis parallels the affected bone (Fig. 36-8A,B). The intramedullary margin has characteristic sharply demarcated, scalloped borders with sclerosis.
Grossly, chondromyxoid fibroma presents as a sharply demarcated, firm, gray-white lobulated mass that is eccentrically located and is always surrounded by intact periosteum.
Microscopically, chondromyxoid fibroma shows a pseudolobulated architecture with myxomatous and chondroid areas separated by hypercellular tissue (Fig. 36-9A,B). The hypercellular component is composed of mononuclear cells with scattered multinucleated giant cells, and shows an overall resemblance to chondroblastoma. Immature myxoid mesenchymal tissue, which creates a pseudolobulated pattern, contains stellate or oval cells that may present some degree of hyperchromasia. Foci of early primitive chondroid differentiation may be present (Bleiweiss and Klein, 1990).
Approximately 30% of chondromyxoid fibromas, especially these located in the small bones of the acral skeleton, show focal features of nuclear atypia. If the microscopic features are analyzed without correlation with clinical and radiologic presentations, they may be misinterpreted as myxoid chondrosarcoma (Wu et al, 1998).
Cytology
Fine-needle aspirates of chondromyxoid fibroma typically show ill-defined lakes of myxoid material with scattered, isolated, primitive mesenchymal cells and occasional multinucleated giant cells (Fig. 36-9C). Less frequently, three-dimensional hypercellular clusters of loosely arranged cells, corresponding to more cellular chondroblastoma-like areas, are present (Fig. 36-9D). In general, a cytologic diagnosis of chondromyxoid fibroma can be established when the microscopic features are correlated with a characteristic radiologic presentation in its typical location. If the chondromyxoid fibroma involves less typical anatomic sites and the radiographic presentation is equivocal, it is usually cytologically diagnosed as an unclassified benign myxoid lesion.
Chondroblastoma
Pathology and Histology
Chondroblastoma is a distinct benign cartilage tumor composed of mononuclear chondroblastic cells that focally produce
immature cartilaginous matrix (Dahlin and Ivins, 1972). Chondroblastomas are rare lesions, accounting for only about 1% of all primary bone tumors. They have a predilection for the epiphyses of the major long tubular bones. The distal femoral and proximal tibial epiphyses, followed by the proximal humerus and proximal femur, are the most frequently involved sites. Epiphyseal-equivalent sites of flat bones, such as the periacetabular region in the pelvis, may also be involved. Most chondroblastomas are diagnosed in skeletally immature patients in their second decade of life. Approximately 10% of these lesions are diagnosed in patients older than 50 years (Turcotte et al, 1993a).
immature cartilaginous matrix (Dahlin and Ivins, 1972). Chondroblastomas are rare lesions, accounting for only about 1% of all primary bone tumors. They have a predilection for the epiphyses of the major long tubular bones. The distal femoral and proximal tibial epiphyses, followed by the proximal humerus and proximal femur, are the most frequently involved sites. Epiphyseal-equivalent sites of flat bones, such as the periacetabular region in the pelvis, may also be involved. Most chondroblastomas are diagnosed in skeletally immature patients in their second decade of life. Approximately 10% of these lesions are diagnosed in patients older than 50 years (Turcotte et al, 1993a).
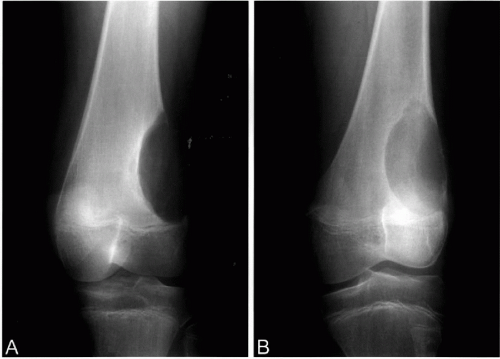 Figure 36-8 Chondromyxoid fibroma: radiographic features. Plain radiographs of a large chondromyxoid fibroma of the distal femoral metaphysis. The lesion is completely radiolucent, sharply demarcated by a sclerotic margin, and eccentric in location. |
Chondroblastoma may recur after simple curettage, and may produce benign lung implants that can be successfully treated by surgery. In extremely rare instances, long-lasting chondroblastomas may progress to a high-grade sarcomatoid malignancy.
Radiographically, chondroblastomas are benign-appearing oval or round lytic lesions located in the epiphysis, with a sharply demarcated sclerotic margin (Fig. 36-10A-D). Larger tumors may expand the bone contour but they do not erode the overlying cortex (Bloem and Mulder, 1985




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
