TABLE 8.1 Common Spatial Distribution of Bone Tumors in Long Bones | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
grounds. Malignant bone sarcomas carry a significant risk of distant metastases, ranging from 20% to 100% depending on the histotype and grade.
TABLE 8.2 Tumor Syndromes in Which Bone Tumors Are Frequently Encountered | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
histologic subtypes are considered high grade (mesenchymal and dedifferentiated chondrosarcoma) or low grade (clear cell chondrosarcoma).
TABLE 8.3 Specific Genetic Changes in Bone Tumors That Can Be Used in Diagnosis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 8.4 Bone Sarcoma Grading | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
discussed earlier. Therefore, a grade 1 or 2 tumor in the AJCC system is equivalent to a stage I tumor in the MSTS system; grade 3 or 4 is equivalent to MSTS stage II. In addition, the TNM staging system includes histologic subtype, size, continuity, and grade as well as local and distant spread to estimate the prognosis of the patient (Table 8.6). Lymph node metastases are, however, rare in bone sarcomas.
TABLE 8.5 Musculoskeletal Tumor Society Staging System for Bone Tumors | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 8.6 TNM Staging System for Bone Tumors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
the presence of matrix mineralization in the form of “popcorn” calcifications is most typical. The edge of the lesion is most often lobular and sharply circumscribed. Histologically, enchondroma consists of lobular, relatively cell-poor hyaline cartilage, often demarcated by a zone of reactive bone formation (encasement) (Fig. 8.7). The chondrocytes have nuclei with condensed chromatin and are evenly dispersed. Binucleated cells are infrequent and mitoses are absent. Occasionally, degenerative features such as ischemic necrosis or calcifications are found (Fig. 8.8). The distinction between an enchondroma and atypical cartilaginous tumor/chondrosarcoma grade 1 can be difficult radiologically (Fig. 8.9) (17). Moreover, this distinction is also difficult on histology (18), causing high interob-server variability (19,20). Mucomyxoid matrix degeneration and entrapment of preexisting host bone are most predictive
of atypical cartilaginous tumor/chondrosarcoma grade 1 (Fig. 8.10) (20). Immunohistochemistry or molecular diagnostics cannot help in this differential diagnosis. Radiologic presentation, localization, and age should also be taken into account. For instance, in the phalanges, the matrix of enchondromas can contain more myxoid features, and the lesion may be more cellular. At this location, the main characteristics of malignancy are the presence of mitoses, cortical breakthrough, and soft tissue involvement (16). Malignant transformation of enchondromas is thought to be extremely rare (<1%) (13). Multiple enchondromas are seen in the context of nonhereditary Ollier disease (multiple enchondromas with a unilateral predominance) (Fig. 8.11) and Maffucci syndrome (multiple enchondromas combined with [spindle cell] hemangiomas) (21) caused by somatic mosaic mutations in the IDH1 or IDH2 gene (22,23). In these conditions, the risk of malignant
transformation is increased to 18% to 46%, depending on the extent and localization of the lesions (24).
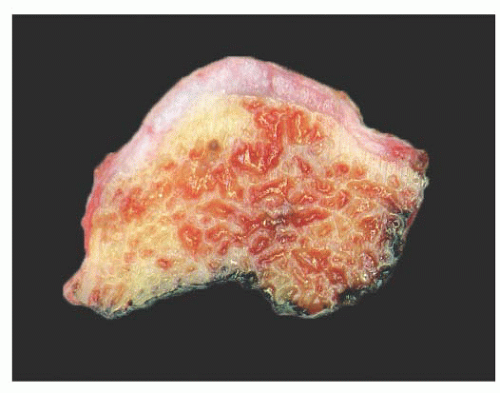 FIGURE 8.1 Gross specimen of an osteochondroma. The lesion is sessile formed by a stalk of medullary bone covered by a smooth, pale, blue-gray cartilage cap that has a thickness of less than 1 cm. |
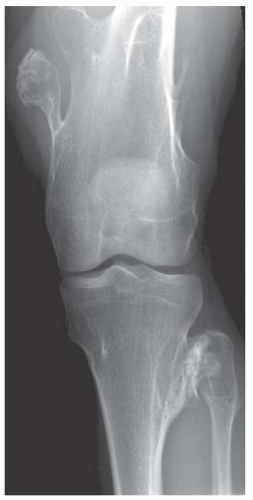 FIGURE 8.2 Multiple osteochondromas. Multiple bony lesions around the knee, projecting away from the joint. |
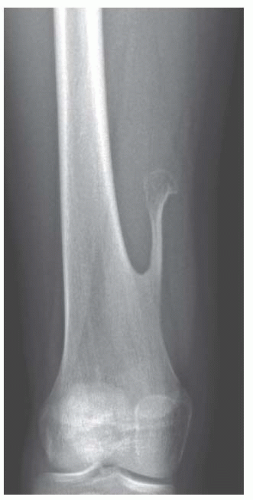 FIGURE 8.3 Osteochondroma of the femur. Note that the medullary cavity of the lesion is continuous with the medullary cavity of the bone. |
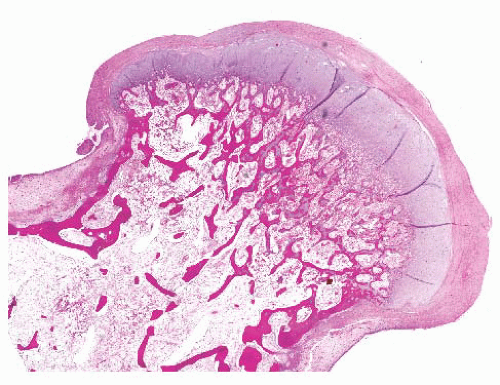 FIGURE 8.4 Osteochondroma. The tumor consists of three layers: perichondrium, cartilage, and bone. |
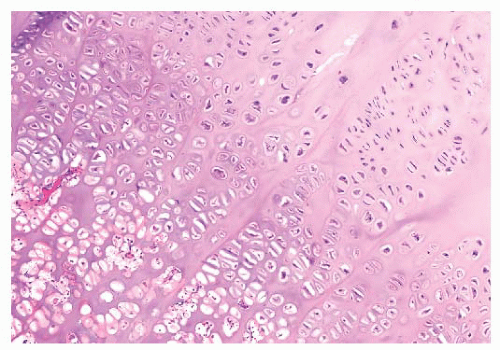 FIGURE 8.5 Osteochondroma. The cartilage cap in osteochondroma usually displays a growth plate-like columnar arrangement of chondrocytes. Endochondral ossification is present at the cartilagebone interface. |
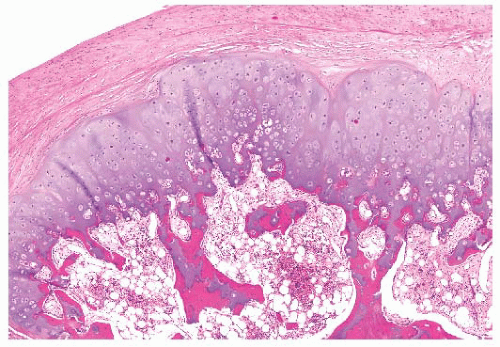 FIGURE 8.6 Osteochondroma. The cartilage cap contains small chondrocytes as well as hypertrophic cartilage cells. Trabecular bone is seen at the base of the cartilage cap. |
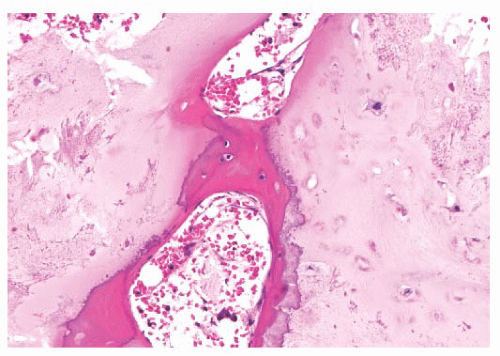 FIGURE 8.7 Enchondroma. The lesion consists of lobular, relatively cell-poor hyaline cartilage, often demarcated by a zone of reactive bone formation (encasement) indicating slow growth. |
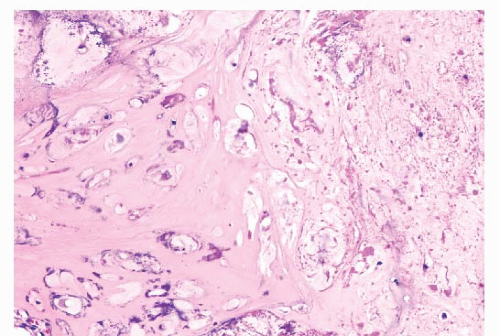 FIGURE 8.8 Enchondroma. The cells have nuclei with condensed chromatin and are irregularly dispersed. Binucleated cells are infrequent and mitoses are absent. Occasionally, necrosis and irregular calcifications are observed. |
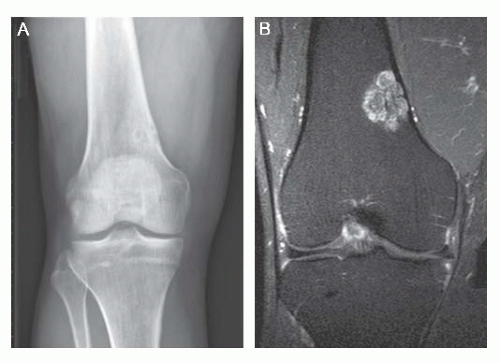 FIGURE 8.9 Atypical cartilaginous tumor/chondrosarcoma grade 1 of the distal femur. (A) The lesion is eccentric with sharp sclerotic margin and areas of calcification. (B) Erosion of the cortex can be seen (endosteal scalloping). The distinction between enchon-droma and atypical cartilaginous tumor/chondrosarcoma grade 1 is very difficult at radiography. |
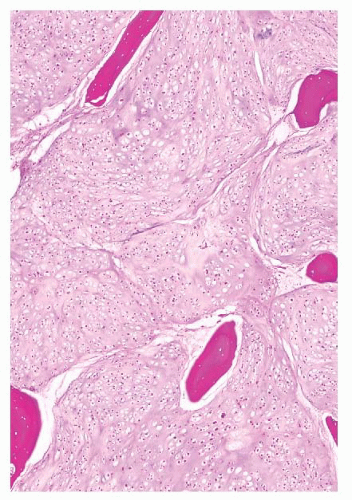 FIGURE 8.10 Chondrosarcoma. Entrapment of preexisting host bone by the cartilaginous neoplasm indicating invasive growth. |
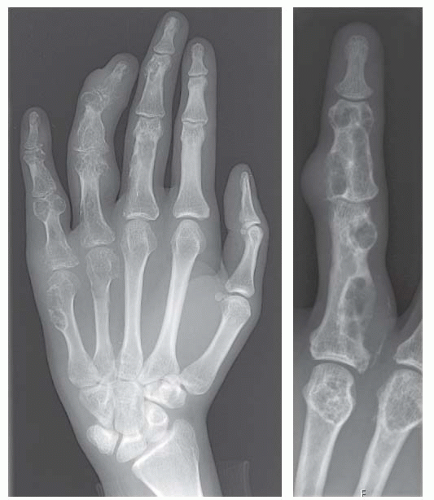 FIGURE 8.11 Ollier disease. Conventional radiograph of the hand demonstrating multiple enchondromas affecting multiple bones. |
is typically located at the distal phalanx, whereas BPOP is not located at that site. In contrast to osteochondroma, which is extremely rare on the digits, the cartilaginous matrix is highly fibrillary. Subungual exostosis is characterized by a recurrent chromosomal translocation t(X;6), rearranging the COL12A1 and COL4A5 genes in chromosome bands 6q13-14 and Xq22 (33), upregulating expression of the IRS4 gene (34).
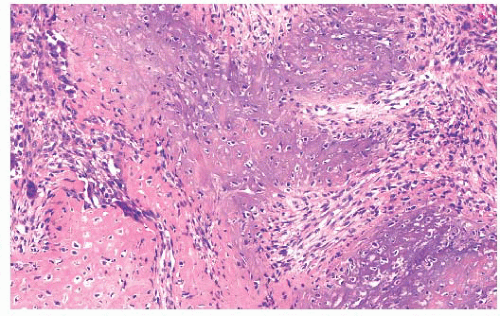 FIGURE 8.12 Bizarre parosteal osteochondromatous proliferation of bone. The lesion is composed of a proliferation of cartilage, which matures into bone, and bone-forming cells in a fibroblastic stroma. Bluish calcification is seen. |
chondromyxoid fibroma express S100, and the cells at the periphery of the lobules are myofibroblastic, expressing MSA and SMA. Chromosome 6 aberrations are frequent (38). These variable rearrangements were all shown to target the GRM1 gene, as they all result in upregulation of GRM1 (39). High GRM1 expression was shown to be specific for chondromyxoid fibroma, suggesting that aberrant glutamate signalling is involved in its histogenesis (39).
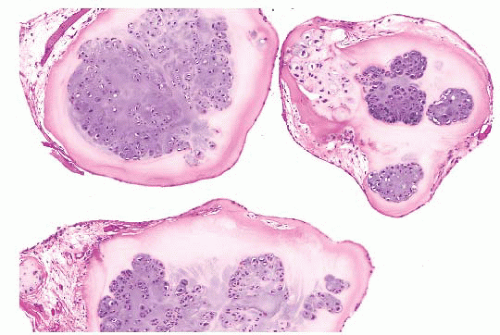 FIGURE 8.13 Synovial chondromatosis. Nodules of cartilage are surrounded by a thin layer of synovium. Chondrocytes are clustered together in nests. |
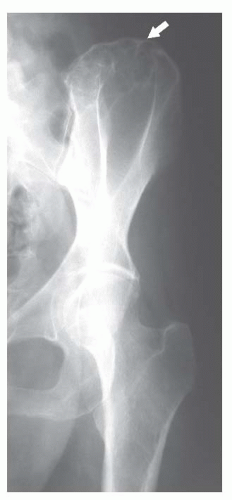 FIGURE 8.14 Chondromyxoid fibroma of the iliac wing. The lesion is seen as an eccentric (arrow), well-circumscribed lytic defect, with a “soap-bubble” appearance. |
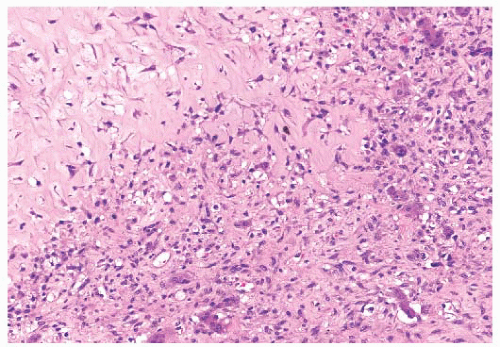 FIGURE 8.15 Chondromyxoid fibroma. Lobules of myxoid or chondroid matrix with numerous spindle-shaped or stellate cells (left) are seen, with increased cellularity at the edge of the lobules (right) where cells are more rounded and admixed with multinucleated osteoclastic giant cells. |
Immunohistochemistry is not particularly helpful in this distinction, although recently, the expression of DOG1 by clusters of cells in chondroblastoma has been reported (41). Very recently, a consistent mutation in H3F3B (p.Lys36Met), which is one of the two genes for histone H3.3, was found in chondroblastoma (42). Interestingly, giant cell tumor of bone harbors alterations in H3F3A, the other gene for histone H3.3 (42).
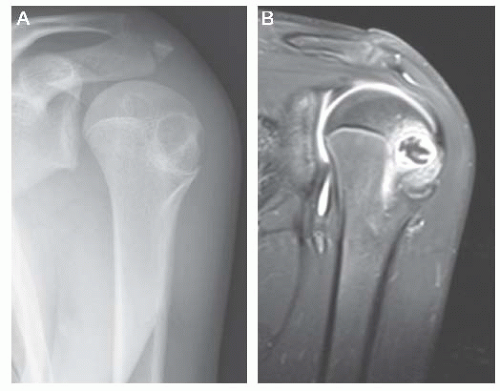 FIGURE 8.16 Chondroblastoma of the proximal humerus. (A) The lesion is typically located at the epiphysis, is well circumscribed, and has a sclerotic rim. (B) Extension of the chondroblastoma across the physis and reactive edema visible on MRI. |
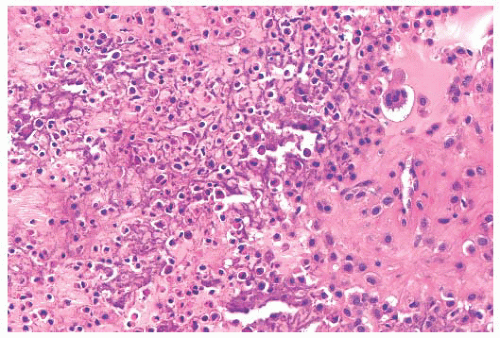 FIGURE 8.17 Chondroblastoma. Immature, rounded or polygonal chondroblast-like cells with sharp cell borders embedded in a chondroid-like matrix and admixed with multinucleated osteoclast-like giant cells. |
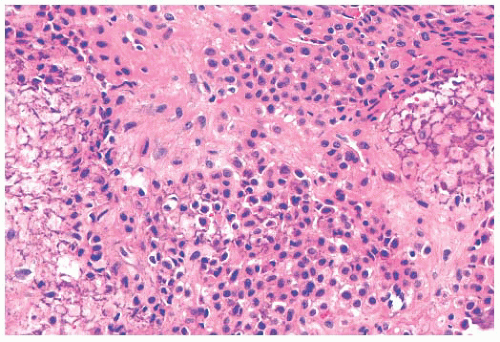 FIGURE 8.18 Chondroblastoma. Lacy, “chicken wire” calcification occurs within the matrix (right). |
they occur in enchondromas and therefore do not distinguish between enchondroma and chondrosarcoma (23,28). The (epi)genetic events causing malignant transformation of enchondroma to chondrosarcoma are so far unknown, although upon progression, chondrosarcomas become aneuploid and karyotypes become complex with increasing histologic grade, with aberrations in the p53 and Rb pathways.
TABLE 8.7 Clinicopathologic Features of the Different Types of Chondrosarcoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
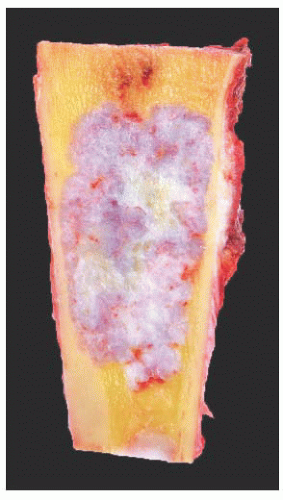 FIGURE 8.19 Gross specimen of chondrosarcoma. The tumor occupies much of the medulla and has the pearly white and lobular appearance of cartilage. Note the endosteal scalloping. |
select several areas to be submitted for histology. The tumor needs to be graded based on those areas demonstrating the highest grade. Also, histologic grading has been shown to be subject to interobserver variability (19,20). An absolute correlation between histology and biologic behavior is lacking. This is especially true for the chondrosarcomas of the phalanges, in which the histology can be highly malignant but the behavior indolent. Grading of phalangeal chondrosarcomas therefore does not seem useful (16).
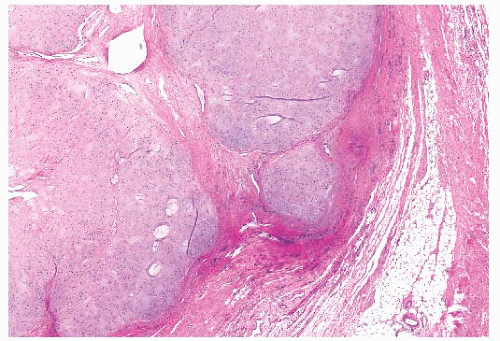 FIGURE 8.20 Secondary peripheral chondrosarcoma. Lobules of tumor cartilage separated by fibrous tissue are seen invading soft tissue. |
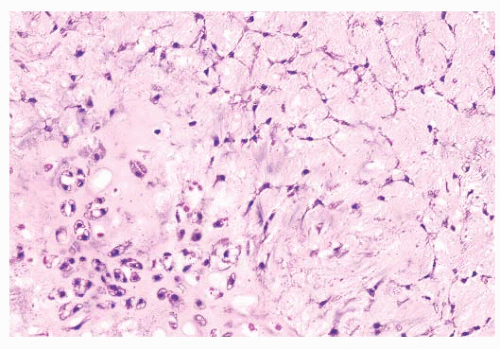 FIGURE 8.21 Grade 2 chondrosarcoma. The cellularity is increased and myxoid change of the matrix can be seen (right). |
Within periosteal osteosarcomas, by definition, direct deposition of osteoid by tumor cells is seen. Moreover, the cartilaginous areas in periosteal osteosarcoma display considerable nuclear pleomorphism, whereas this is usually limited in chondrosarcoma.
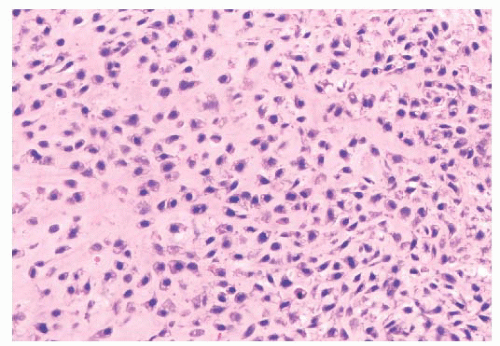 FIGURE 8.22 Grade 3 chondrosarcoma. The neoplasm contains highly cellular areas with spindle cell change. |
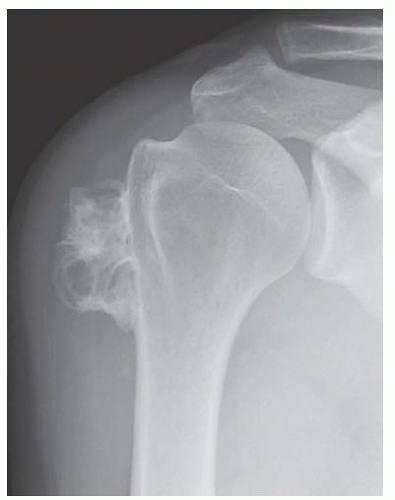 FIGURE 8.23 Periosteal chondrosarcoma of the proximal humerus. A well-circumscribed cartilage mass on the bone surface producing erosion of the underlying cortex. |
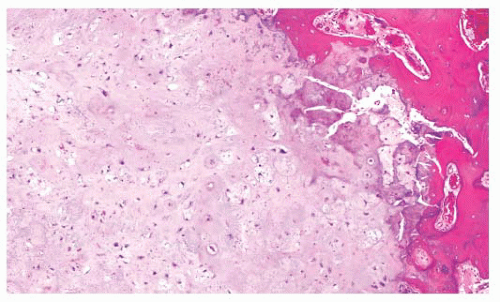 FIGURE 8.24 Periosteal chondrosarcoma. Low- and highly cellular areas, myxoid change of the matrix, and cortical invasion (right). |
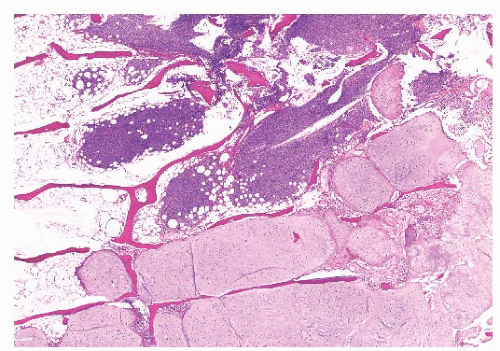 FIGURE 8.25 Dedifferentiated chondrosarcoma. There is an abrupt transition between the conventional low-grade cartilaginous component (lower half) and the high-grade spindle cell sarcoma (upper half). Note the permeative growth pattern. |
small cell component is positive for SOX9 and variably for CD99 and can show expression of desmin. The undifferentiated small cell areas may morphologically resemble Ewing sarcoma, and both tumors contain cytoplasmic glycogen (57). However, by definition, cartilage is absent in Ewing sarcoma. Furthermore, small cell osteosarcoma and dedifferentiated chondrosarcoma should also be considered in the differential diagnosis. Irregular fine trabecular deposition of osteoid, characteristic of small cell osteosarcoma, and a sharp demarcation between the two components, characteristic of dedifferentiated chondrosarcoma, are absent in mesenchymal chondrosarcoma (55). A recurrent HEY1-NCOA2 fusion has been identified in mesenchymal chondrosarcoma (58). When the cellular, undifferentiated component predominates, the tumors can be sensitive to chemotherapy and radiation (56).
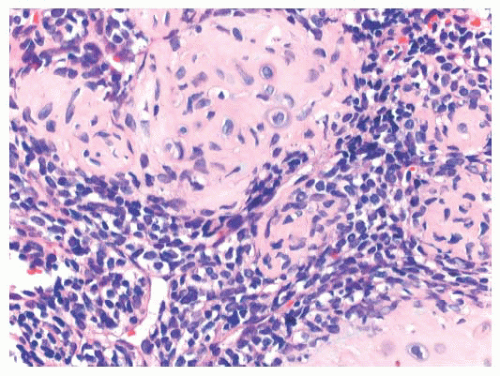 FIGURE 8.26 Mesenchymal chondrosarcoma. The neoplasm is formed by areas with more or less differentiated cartilage mixed with vascular, cellular areas containing undifferentiated small spindleshaped or round cells with scant or no cytoplasm. |
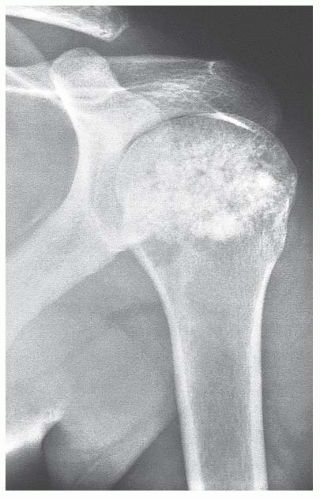 FIGURE 8.27 Clear cell chondrosarcoma of the proximal humerus. The lesion is heavily mineralized, extends to the end of the bone, and appears well circumscribed.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|