Fig. 21.1.
Myositis ossificans (gross) forming a circumscribed mass with a bony shell and a hemorrhagic fibrous center (A). Myositis ossificans (micro) at frozen section a well developed “zonation” is identified (B). Mature new bone formation in a patient with myositis ossificans of the soft tissues of the thigh (C–D).
♦
The central fibroblastic repair reaction can be alarming and sometimes mistaken for a malignant sarcoma if taken out of context without any attention being paid to the low-power impression of zonation (Fig. 21.1B)
Differential Diagnosis
♦
Fibrodysplasia progressiva (previously called myositis ossificans progressiva): this is an entirely different and progressive condition that is genetic
♦
Bone-forming tumors (such as osteosarcoma): can be differentiated since there is no peripheral maturation (zonation) in tumors. Additionally, osteosarcoma has a hypercellular spindle cell proliferation with considerable cytological atypia and sometimes sheets of atypical osteoblasts
Reflex Sympathetic Dystrophy (Algodystrophy or Sudeck Osteodystrophy)
Definition
♦
A condition of severe regional, patchy osteopenia following trauma that is almost always accompanied by pain, redness, and swelling of the affected portion of body (generally the limbs)
Clinical
♦
It is associated with trophic skin changes, edema of the extremity, and psychological disturbances
♦
The radiological differential diagnosis includes other forms of osteopenia such as bone marrow edema, transient osteoporosis, migratory osteolysis, and idiopathic regional osteoporosis. The clinical findings of pain and sympathetic nervous system involvement can help differentiate these conditions
Microscopic
♦
The changes are not very specific. There is a proliferation of fibroblasts within the marrow space
♦
In some cases, bone necrosis and new bone formation may be seen
Osteonecrosis (Avascular, Aseptic, Ischemic Necrosis)
Definition
♦
The in situ death of bone, presumably from one or more vascular insults. By definition, necrosis of bone occurring as a result of infection is excluded (Fig. 21.2A). The term osteonecrosis includes instances resulting from insults such as radiation and is a more encompassing term than avascular necrosis although the morphological features are almost identical
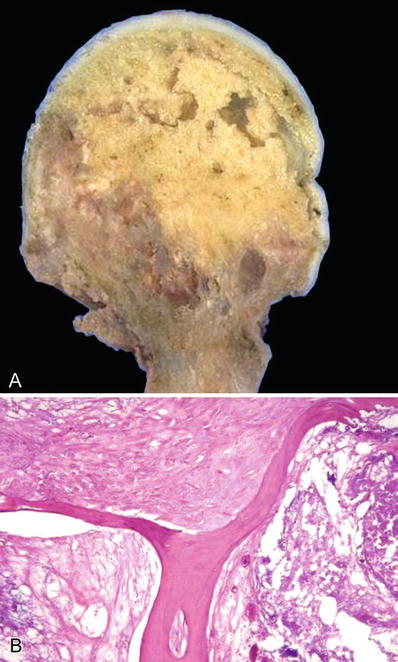
Fig. 21.2.
Avascular necrosis of the femoral head (gross) (A). The photograph shows the infarcted bone beginning to separate from the adjacent bone and the overlying cartilage (A). The trabeculae are anucleate, and the adjacent marrow shows necrosis and fibrosis (B).
Clinical
♦
There are two main forms:
Medullary infarction (marrow cavity and trabecular bone). This form is often silent
Corticomedullary infarction (cortex also involved). This form is often painful
♦
Conditions predisposing to osteonecrosis:
Trauma, infection, and fatigue fractures
Alcohol abuse
Dysbarism
Gaucher disease
Connective tissue disorders
Vasculitis
Hemoglobinopathies and coagulopathies
Radiation injury
Corticosteroid therapy
Pregnancy
Aging
Gout
Pancreatitis
Childhood osteochondritides such as Perthe, Keinbock, Sever, Kohler, Larsen, Blount, or Panner diseases (the pathogenesis of these is uncertain, probably avascularity, but the morphological features are those of osteonecrosis)
♦
Osteonecrosis is suspected on the basis of clinical history, physical findings, X-ray, and other scanning techniques. Magnetic resonance imaging (MRI) is especially sensitive and helpful in the diagnosis
♦
Diagnosis is confirmed by biopsy
Microscopic
♦
The hallmark of osteonecrosis is the finding of anucleate bone (with empty lacunae) (Fig. 21.2B). Surrounding this is reactive hyperemia in the early course. This is followed by a fibrovascular proliferation adjacent to the necrotic bone. The necrotic bone is then walled off, in a fashion similar to sequestrum formation in osteomyelitis
♦
Revascularization of dead bone occurs in a few weeks
♦
In a few days, cutting cones of osteoclasts arrive, carrying blood vessels. They enter the dead bone and remove it gradually by osteoclastic resorption
♦
At the same time, osteoblasts lay down new bone on top of the necrotic fragment. The laying down of new (woven) bone on top of necrotic trabeculae is quite characteristic
♦
The next stage is unpredictable, depending on the site and extent of damage:
May culminate in restitution (complete healing)
May lead to the relentless continuation of the repair process that damages the integrity of bone. This then leads to fatigue fractures, collapse of the subchondral bone , cartilage disintegration, and joint deformity (secondary osteoarthritis)
Osteochondritis “Juvenilis”
♦
Refers to the earlier set of eponymic names for avascular necrosis occurring in certain bones
♦
Includes conditions such as Perthe, Keinbock, Sever, Kohler, Larsen, Blount, or Panner diseases
♦
With the exception of Perthe disease (which may be associated with constitutional skeletal abnormalities), these conditions are similar to the osteonecrosis occurring in the noneponymic locations
BONE INFECTIONS
Pyogenic Osteomyelitis
Acute Osteomyelitis
Clinical
♦
Infections can reach the bone by:
Hematogenous route
•
From an antecedent focus of infection elsewhere. This is the most common form of osteomyelitis, especially in children
•
Most cases of hematogenous osteomyelitis occur in the metaphyses of long tubular bones in children
Direct extension
•
Infection can also reach bone by direct extension, for example, from an infected wound or a deep chronic ulcer overlying the bone. Often, this form of osteomyelitis is seen in association with trauma (compound fractures)
♦
Commonly encountered organisms
Staphylococcus aureus (most common)
Escherichia coli, Klebsiella, and Pseudomonas species
Salmonella (seen in patients with sickle cell anemia in the form of a cortically based osteomyelitis)
Treponema, gram negative rods, Streptococci, Haemophilus influenzae, and Listeria species (occur in neonates)
Pseudomonas infections (seen in addicts abusing intravenous street drugs)
Macroscopic
♦
The infected region of bone is often like an abscess filled with necropurulent exudate and surrounded by intensely hyperemic fibrovascular tissue . The pus is in a rigid confined space (bone) and so exerts pressure on the surrounding small vessels. This may result in additional ischemic damage to the bone
♦
The exudate follows the path of least resistance and exits through the Volkmann canals into the subperiosteal space
♦
In the neonatal age group , the Sharpey fibers are less well developed; thus, considerable subperiosteal spread may occur
♦
The pus may also track along the medullary cavity. This can become quite extensive in some cases
♦
In severe cases, the combination of subperiosteal and intramedullary spread can cause the entire diaphysis to become necrotic, forming a “ring sequestrum”
Microscopic
♦
There is bone destruction surrounded by polymorphonuclear leukocytes and cell debris. The necrotic bone is called a sequestrum
Chronic Osteomyelitis
Clinical
♦
Although de novo cases also occur, the vast majority of chronic infection results from unresolved acute osteomyelitis
♦
Chronic osteomyelitis follows a protracted course, interspersed with acute exacerbations
♦
Because of the low levels of causative organisms, cultures may be negative
♦
Common organisms include:
Staphylococcus aureus
Streptococci (group A is more frequent)
Klebsiella (may cause extensive bone damage)
Aerobacter, Proteus, Brucella, Staphylococcus epidermidis, and Bacteroides
Microscopic
♦
Necrotic bone in a chronic inflammatory background
♦
When the necrotic fragment separates from the adjacent tissue, it is known as a sequestrum
Separation of the sequestrum generally takes months to complete
The sequestrated bone is often of cortical origin
♦
The bone surrounding a focus of chronic osteomyelitis is often dense and is referred to as the “involucrum ”
The involucrum is often of periosteal origin
The involucrum frequently has several openings of “cloacae” through which exudate, bone debris, and sequestra exit and pass through sinus tracts to the surface (Fig. 21.3A)
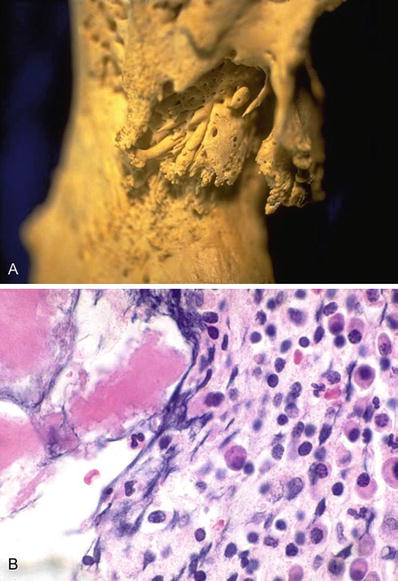
Fig. 21.3.
Chronic osteomyelitis showing bone thickening and cloaca/sinus formation (A). Chronic osteomyelitis may show several plasma cells and have to be differentiated from a plasma cell dyscrasia or myeloma (B).
Constant destruction of the neighboring soft tissues leads to scarring and squamous metaplasia of the sinus tract
♦
The microscopic diagnosis of chronic osteomyelitis can be difficult since in some cases, the inflammatory infiltrate may be sparse and may mimic the normal elements of the marrow space. Alternatively, in other cases, it may be mistaken for a hematological malignancy (Fig. 21.3B)
♦
Microscopic recognition of the sequestrum is helpful for the diagnosis of infection
The sequestrum is recognized by virtue of its anucleate nature; often the edges are jagged (because of the action of proteolytic enzymes and osteoclastic action)
Overdecalcification of bone during processing can cause the bone to become artificially “anucleate,” making the recognition of dead bone difficult
Differential Diagnosis
♦
Normal marrow
Preservation of normal fat pattern of marrow
Lack of a fibrous background
Sclerosing Osteitis (Garre Osteomyelitis)
Definition
♦
Sclerosing osteitis is gradual, usually unilateral (occasionally bilateral and symmetrical) bony sclerosis, often associated with pain. It is speculated to be a form of chronic osteomyelitis
Clinical
♦
Constitutional symptoms are infrequent
♦
Usually affects children
♦
Most frequent sites include the tibia, jaw, and clavicle (and pubis in adults)
Microscopic
♦
The biopsy findings are nonspecific, and often there is no significant inflammation
♦
Culture results are often negative
Chronic Multifocal Recurrent Osteomyelitis
Clinical
♦
Patients present with recurrent episodes of bone pain, erythema, and swelling
♦
X-rays and bone scans are consistent with osteomyelitis
♦
The signs and symptoms usually resolve with time. Antimicrobial therapy is not helpful
Microscopic
♦
Bone biopsies confirm the suspicion of osteomyelitis
♦
Organisms are usually not isolated
♦
May be related to the seronegative spondyloarthropathies
Tubercular Osteomyelitis
Clinical
♦
Tubercular osteomyelitis is generally more frequent in children although there is a wide age range, especially for tubercular spondylitis (Pott spine), which often afflicts adults
♦
In the long tubular bones, the disease usually has a metaphyseal location; in adults, the vertebrae (or rarely epiphysis of long bones) can be involved
Pathology
♦
May affect the bone, the joints, or (frequently) both (Fig. 21.4A)
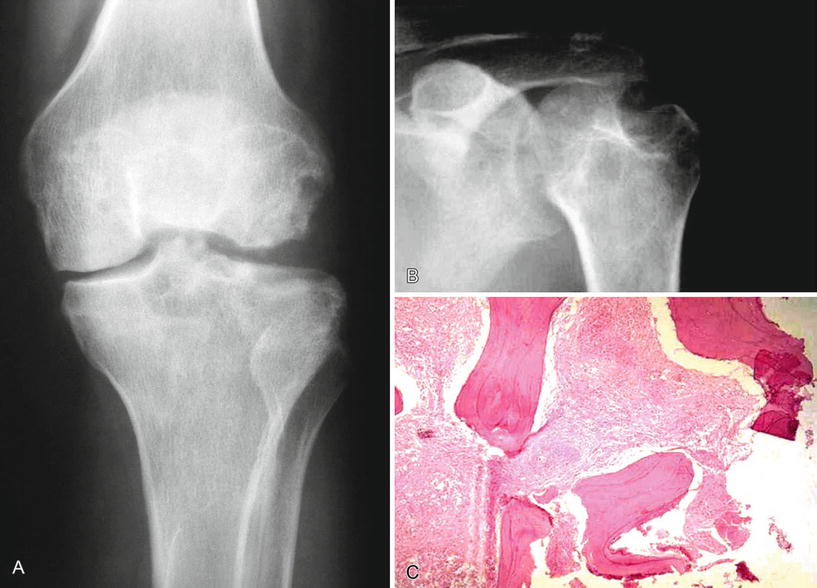
Fig. 21.4.
Tuberculosis of the knee showing multiple lucencies and an erosion of the femoral condyle (A) Caries sicca – a “dry or nonexudative” form of tuberculosis involving the shoulder joint (B). Granulomatous inflammation in the intertrabecular spaces (C).
♦
Often occurs by hematogenous spread
♦
Is usually associated with a reactivation of a preexisting primary focus in the lung
♦
The emergence of multidrug-resistant tuberculosis and the AIDS epidemic has reemphasized the importance of mycobacterial infections
Microscopic
♦
The disease has traditionally been divided into the “dry or granular” and the “exudative” types (Fig. 21.4B)
♦
The hallmark of the infection is the finding of granulomas. Quite often, these are of the necrotizing type (Fig. 21.4C)
♦
Special stains for acid-fast organisms may be positive (but in many cases, negative also). Cultures are more frequently positive than histological stains
Fungal Osteomyelitis
♦
Fungal infection of bone is uncommon
♦
More common infections include:
Blastomycosis
Coccidioidomycosis
Actinomycosis
Maduromycosis
Sporotrichosis
♦
These infections often involve the hands, feet, or craniofacial skeleton
ARTHRITIDES
Osteoarthritis (OA)
♦
Also termed degenerative joint disease
♦
Generally refers to a failure of diarthrodial (movable, synovium-lined) joint
♦
There are two forms:
Idiopathic (primary) OA
•
Most common
•
No known predisposing factor
Secondary OA
•
Attributed to an underlying cause (developmental disorders, metabolic or endocrine conditions, crystal deposition, trauma, infection, avascular necrosis, neuropathic disease, etc)
•
Pathologically indistinguishable from idiopathic form
Microscopic
♦
The most striking changes are seen in load-bearing areas of the articular cartilage
♦
In the very early stage, the cartilage is thicker than normal. This stage is not often seen in surgical pathology material
♦
The joint surface eventually becomes thinner, and the cartilage softens with progression of the disease
♦
The integrity of the surface is breached, and vertical clefts develop with fibrillation (Fig. 21.5)
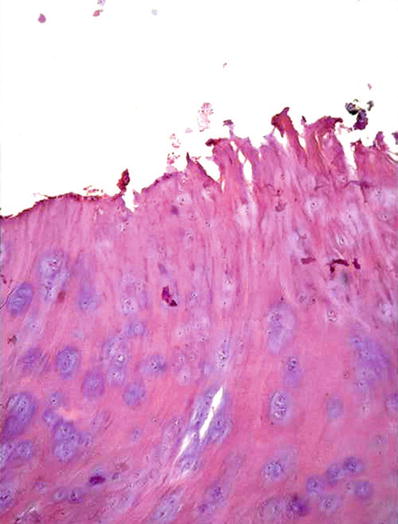
Fig. 21.5.
Osteoarthritis with clefting of the articular cartilage.
♦
Areas of fibrocartilaginous repair may develop
♦
The articular cartilage is metabolically active, and the chondrocytes replicate, forming clusters (termed “cloning”). Later, the cartilage becomes hypocellular
♦
Fractures of the subchondral plate can occur, or there can be osteoarthritic cyst formation in the subchondral region
♦
Remodeling and hypertrophy of bone are also major features:
Appositional bone growth occurs in the subchondral region
The abraded bone under a cartilage ulcer may resemble ivory wood (termed “eburnation”)
♦
Growth of cartilage and bone at the joint margin leads to osteophytes (spurs)
♦
Osteophytes alter the contour of the joint, restricting movement
♦
Thickening of the joint capsule along with chronic synovitis further restricts joint movement
Neuropathic Arthropathy (Charcot Joints)
Clinical
♦
An extremely destructive joint disorder in patients with neurosyphilis, diabetes, or other causes of damaged sensory innervation to the joint
♦
With the decline of tertiary syphilis, other causes of neuropathic joints have now become more important, such as:
Diabetes
Syringomyelia
Amyloidosis
Alcoholic neuropathy
Leprosy
Microscopic
♦
There is marked fragmentation of the joint surface with extensive detritic synovitis resulting from particles of bone and cartilage embedded in the synovium
Crystal Arthropathies
Pathogenesis
♦
Crystal arthropathy can be caused by endogenous (monosodium urate [MSU], calcium pyrophosphate) and exogenous (corticosteroid ester crystals, talc, polyethylene, methyl-methacrylate) crystal deposition, producing disease by triggering a cascade that results in cytokine-mediated cartilage destruction
Gout and Gouty Arthritis
Clinical
♦
Gout is the common end point of a group of disorders that produces hyperuricemia
♦
Marked by transient attacks of acute arthritis initiated by the crystallization of urates within and around joints
♦
Deposition of masses of urates in joints and other sites, creating tophi
♦
Factors that may result in the conversion of hyperuricemia into primary gout:
Age (gout rarely appears before age 20–30 years)
Duration of the hyperuricemia
Genetic predisposition
•
X-linked abnormalities of hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
•
Multifactorial inheritance
Heavy alcohol consumption
Obesity
Thiazide diuretics
♦
Gout can result in:
Acute arthritis
Chronic tophaceous arthritis
Gouty nephropathy
Tophi in various sites
Microscopic
♦
Characteristic needlelike crystals that are negatively birefringent under polarized light
Calcium Pyrophosphate Crystal Deposition Disease (Pseudogout, Chondrocalcinosis)
♦
Three types:
Sporadic
Hereditary
Secondary types linked to:
•
Hyperparathyroidism
•
Hemochromatosis
•
Hypomagnesemia
•
Hypothyroidism
•
Ochronosis
•
Diabetes
♦
More common at age 50 or above
♦
Joint involvement may mimic OA or rheumatoid arthritis (RA)
♦
Pathogenesis is uncertain:
Altered activity of the cartilage matrix enzyme produces and degrades pyrophosphate, resulting in its accumulation and eventual crystallization
The crystals first develop in the articular matrix, menisci, and intervertebral discs
Rheumatoid Arthritis
Clinical
♦
A systemic disease that manifests in the joints as a synovial lesion
♦
Autoimmune inflammatory process that primarily targets the synovial tissue
Humoral and cellular immune response is involved
Microscopic
♦
Nonsuppurative chronic inflammation in the capsule of the joint
Hypertrophy and hyperplasia of the synovial cells resulting grossly in a papillary pattern on the surface of the synovium
Later, a pannus forms
•
A combination of proliferating mesenchymal cells and granulation tissue starts at the periphery of a joint, subsequently destroying articular cartilage
METABOLIC BONE DISEASES
Osteoporosis
Definition
♦
A condition characterized by a reduced amount of normally mineralized bone
Classification
♦
Postmenopausal (type I)
♦
Age related (type II)
♦
Secondary (accounts for ~5% of cases)
Can be seen in a variety of conditions
•
Osteogenesis imperfecta (OI)
•
Turner syndrome
•
RA
•
Systemic mastocytosis
•
Hyperthyroidism
•
Adrenal disease
•
Steroid or heparin therapy
•
Chronic alcoholism
•
Space travel
Clinical
♦
Higher incidence associated with:
Race (Caucasians > African Americans)
Sex (F > M)
Physical inactivity
Slender body builds
Smoking
Nulliparity
Early menopause
♦
The major clinical symptoms are those of fractures
♦
Common sites of fractures are:
Spinal vertebral crush fractures
Hip fractures
Colle fracture or other fractures of the distal radius
♦
Laboratory investigations include the exclusion of other metabolic diseases by assessing:
Serum calcium
Serum phosphate
Alkaline phosphatase
25-hydroxy and 1,25-dihydroxy-vitamin D
Urinary calcium
♦
Biologic markers that may be useful for assessing bone turnover:
Bone-specific alkaline phosphatase
Serum/urinary osteocalcin
Serum type I collagen extension peptides
Plasma tartrate-resistant acid phosphatase
Urinary levels of hydroxyproline
Urinary pyridinoline cross-links of type I collagen
♦
Pathogenesis
Postmenopausal osteoporosis
•
Associated with increased osteoclastic activity
•
May be initiated or maintained by a variety of bone cytokines, perhaps initiated by RANK and RANK-ligand interaction
•
May be genetically predetermined by the polymorphisms of the Vitamin D receptor gene
Age-related osteoporosis
•
Inefficiency of bone formation in a normal remodeling cycle
•
Less bone is formed than is resorbed with each remodeling cycle throughout life
Microscopic
♦
Cortex
Enlarged Haversian and Volkmann canals tunneled by osteoclasts
The cortex is thinned:
•
Caused by resorption of the subperiosteal and endosteal surfaces
•
Endosteal resorption results in a blurring of the cortical-cancellous border, referred to as “trabeculization ” of the cortex
♦
Trabecular bone
Thinning and perforation of the trabeculae
•
Perforation is an irreversible process, which occurs when an osteoclast resorbs bone all the way through a trabeculum or when two osteoclasts fortuitously located at opposite ends of the trabeculum meet midway
These thin trabeculae seem to “float” in the marrow space
Increased osteoclastic activity may be seen in “high turnover” (postmenopausal) osteoporosis
Rickets and Osteomalacia
Definition
♦
Syndromes (rather than specific disease entities) characterized by a failure of normal mineralization of bone and epiphyseal cartilage
♦
Clinically characterized by bone deformities
♦
In rickets (which occurs in children), bone and epiphyseal cartilage is involved
Causes of Rickets and Osteomalacia
♦
Deficiency states
Diet
Lack of sunlight
♦
Gastrointestinal causes
Gastric resections
Biliary and enteric causes
♦
Renal tubular causes
Hypophosphatemic states
Fanconi syndromes
End-organ defect
Renal tubular acidosis
♦
Unusual causes
Phosphaturic tumors (Fig. 21.6)
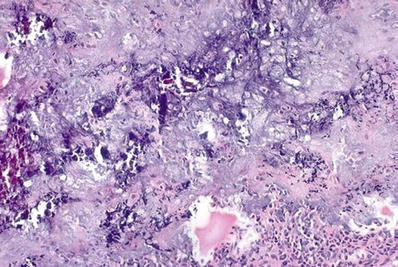
Fig. 21.6.
Mesenchymal phosphaturic tumor (initially called a cartilaginous neoplasm) that caused oncogenic osteomalacia.
Anticonvulsant therapy
♦
Renal osteodystrophy
Renal failure
Pathology
♦
Rickets
A disease of the growing skeleton
Affects the physeal plate and epiphysis of bones of children
♦
Osteomalacia
Occurs in adults, after the growth cartilage has fused and the physis is obliterated
♦
In both instances, there is insufficient ionized calcium or inorganic phosphate (or both) to mineralize the skeleton, leading to less mineralized bone per unit volume of bone
♦
There may in some cases be less bone overall, but more strikingly, the bone that is present fails to mineralize properly
♦
Trabeculae are surrounded by unmineralized osteoid, called “osteoid seams”
Osteoid seams >12.5 μm are virtually diagnostic
Bone histomorphometry has shown that the mineralization lag time is >100 days in rickets/osteomalacia (normal = 80–90 days)
♦
All rachitic syndromes have similar histology, and the individual diagnosis cannot be made on the basis of a bone biopsy
♦
Stains for aluminum may be considered in renal osteodystrophy
♦
In rickets, pressure effects cause deformity at the epiphysis–metaphysis junction, resulting in metaphyseal flaring and a disordered physis
Primary Hyperparathyroidism
♦
This entity was defined in part by von Recklinghausen under the term osteitis fibrosa cystica generalisata
♦
Most often results from a hyperplasia or adenoma (and rarely a carcinoma) of the parathyroid glands:
The diseased gland does not recognize the signal of high serum calcium concentration
There is increased production of 1,25-dihydroxy-vitamin D and parathyroid hormone, increasing absorption of calcium from the gut and bone and preventing its excretion in the kidney
Simultaneously, there is hyperphosphaturia
Serum levels of alkaline phosphatase are frequently high
Microscopic
♦
The bones are characterized by:
Fibrosis of the marrow
Osteoclastic resorption
Osteoblastic rimming on new and often incompletely mineralized lamellar bone trabeculae (narrow osteoid seams)
“Brown tumors ” (Fig. 21.7)
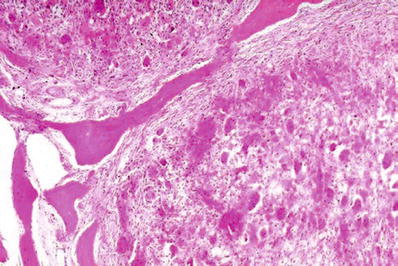
Fig. 21.7.
Brown tumor . There is fibrosis, hemorrhage, and a cluster of osteoclast giant cells.
•
These represent granulation tissue, inflammatory cells, and macrophages containing hemosiderin and giant cell formation
•
There is virtually no bone present in these areas
♦
Occasionally, cystic change may supervene
♦
Increased osteoclastic activity is seen in subperiosteal, intracortical, endosteal, subchondral, and trabecular surfaces
Intracortical resorption is characterized by groups of osteoclasts (known as cutting cones) that tunnel through the cortex, enlarging the Haversian and Volkmann canals
•
These channels are often expanded to 1 mm or more in some cases and may be seen radiographically as lucent lines within the cortex
Endosteal resorption is also visible radiographically as “scalloping” of the cortex at its marrow interface
Subchondral resorption is best seen histologically (and radiographically) at the sacroiliac joint
Paget Disease (Osteitis Deformans)
Clinical
♦
The disease is generally seen in older individuals (unusual before the 4th decade)
♦
There is a slight male predominance
♦
Common among the white population of England, France, Austria, Germany, Australia, New Zealand, and the United States
♦
Rare in Scandinavia, China, Japan, and Africa
♦
May be uni- or multifocal
♦
Most frequent involvement in the axial and the proximal appendicular skeleton
♦
Less frequent involvement in the ribs, fibulae, and bones of the hands and feet
♦
Presenting symptoms :
Pain
Increased width of bone
Weight-bearing bones may be bowed or deformed
May be asymptomatic and discovered incidentally
♦
Complications of disease
Hyperdynamic circulation may lead to a high output cardiac failure
The most common secondary malignancy is osteosarcoma
♦
Radiographically, three phases can be discerned: lytic, mixed, and blastic
Eventually, the disease “burns itself out” in that particular site
♦
Laboratory investigations reflect this increased bone turnover through increased levels of serum alkaline phosphatase and urinary hydroxyproline
Etiology
♦
There is a debate about the role of genetics and infectious viral agents . Current thought appears to be leaning toward the latter. The virus is thought to be a paramyxovirus, similar to measles or respiratory syncytial virus
In situ hybridization studies have localized canine distemper virus (a paramyxovirus) in Pagetic osteoblasts, osteoclasts, and osteocytes
The target of the virus is probably the osteoblast; however, the cell-associated virus particles have only been found in the osteoclasts
Retroviruses can induce the secretion of IL-6 from fibroblasts and macrophages
IL-6 is thought to be important in the pathogenesis of Paget disease and is implicated in the recruitment and activation of osteoclasts
Microscopic
♦
Paget disease is a focal process
♦
The histologic hallmark is mosaic lamellar bone
Characterized by haphazard cement lines (jigsaw-like pattern) (Fig. 21.8)
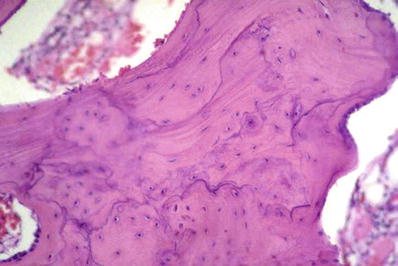
Fig. 21.8.
Mosaic bone of Paget disease.
♦
Lytic phase
Waves of osteoclastic activity
Numerous resorptive pits
♦
Mixed phase
The osteoclasts are admixed with osteoblasts, which line the bone surfaces
The marrow adjacent to the bone becomes replaced by loose, vascularized, connective tissue
The newly formed bone is initially woven bone and is later remodeled into lamellar bone
As the mosaic pattern becomes prominent, cell activity ceases
The fibrovascular tissue is replaced by normal marrow (burnt out phase)
Eventually, the bone becomes larger, with thick, irregular trabeculae and porous cortices
♦
Blastic phase
Characterized by reactive and lamellar bone
Osteopetrosis (Albers–Schönberg Marble Bone Disease)
Definition
♦
A group of hereditary diseases characterized by osteoclast dysfunction resulting in diffuse, symmetric, stone-like, skeletal sclerosis, and abnormally brittle bone
♦
Bisphosphonate-induced osteopetrosis has been reported
Clinical
♦
Several clinical variants have been distinguished:
Autosomal recessive form (“malignant”) (ARO)
•
50% of patients harbor mutations of the ATP 6i gene (TCIRG 1) encoding the osteoclast-specific a3 subunit of the vacuolar H+−ATPase
Autosomal dominant form types 1 and 2
•
ClCN7 gene mutations underlie type 2 ADO. This gene encodes for the type 7 chloride channel located in the osteoclast border membrane
♦
Other genetic abnormalities have been described:
•
Deficiency of osteoclast carbonic anhydrase II , deletions of SRC gene, macrophage colony stimulating factor, etc
♦
Patients have a high postnatal mortality
♦
Survivors have anemia, fractures, and hydrocephalus
♦
Later in life, patients are prone to cranial nerve problems and recurrent infections
♦
Extramedullary hematopoiesis results in hepatosplenomegaly
Microscopic
♦
Bones lack a medullary cavity
♦
The ends of the bones are bulbous (Erlenmeyer flask deformity)
♦
Neural foramina are small, compressing the exiting nerves
♦
There is persistence of the primary spongiosa consisting of ossifying cartilage
♦
Medullary cavity contains little or no hematopoietic elements due to overgrowth and accumulation of the primary spongiosa
♦
Bones are mainly woven and lack remodeling
♦
The numbers of osteoclasts may be normal, increased, or decreased
Osteogenesis Imperfecta
Definition
♦
A heterogeneous group of conditions characterized by bone fragility and mutations in the loci coding for the pro-α1 and pro-α2 collagen chains (COL1A1 on 17q21 and COL1A2 on 7q22.1)
♦
Classification
Type 1
•
Autosomal dominant
•
The less severe form
•
Loose joints and low muscle tone
•
Bone predisposed to fractures
•
Blue/purple tint of the sclera
Type 2
•
Autosomal dominant with new mutation
•
The most severe form
•
Lethal at or shortly after birth due to respiratory failure
•
Numerous fractures and severe bone deformity
Type 3
•
Autosomal dominant with new mutation. Rarely, autosomal recessive forms are observed
•
Bone fractures easily with fractures often present at birth
•
Bone deformity frequent and severe
•
Associated respiratory problems
Type 4
•
Autosomal dominant
•
Severity between types 1 and 3
Microscopic
♦
Amount of osseous tissue is decreased
♦
There may be fibromembranous tissue with foci of bone, especially in the severe forms (this finding is more common in the skull)
♦
In the long bones, the cortices are thin, except at the site of a fracture
♦
Sparse trabeculae in the medullary cavity
♦
The inner (cambium) layer of the periosteum may be prominent
♦
Osteoblasts lining the bone trabeculae tend to be less plump and more spindled than is normal
♦
Osteoclasts are sparse
♦
The growth plates are normal, and ossification is not delayed
♦
The ossification centers may be smaller than normal
♦
The chondrocytes and cartilage matrix appear normal by light microscopy
♦
The teeth are abnormal
The mesodermal component is severely affected and may be deficient
The ectodermal component is usually normal
Mucopolysaccharidoses
Definition
♦
A heterogeneous group of lysosomal storage disorders, associated with glycosaminoglycan excretion in the urine and accumulation in the tissues
♦
Deficiencies in enzymes (acid hydrolases) that degrade the glycosaminoglycans
♦
Cartilage tends to be severely affected
Classification (Several Types and Common Ones)
♦
Type 1 (Hurler syndrome)
Associated with defects in α-l-iduronidase
Results in accumulation of dermatan sulfate
♦
Type 2 (Hunter syndrome)
Associated with defects in l-iduronosulfate sulfatase
Results in accumulation of heparan sulfate
Gaucher Disease
Definition
♦
A group of conditions resulting from different allelic mutations in the structural gene of the enzyme glucocerebrosidase
Biochemical Basis
♦
The gene of glucocerebrosidase is located on chromosome 1q21
♦
The enzyme cleaves glucose from ceramide
Clinical
♦
Glucocerebroside accumulates in phagocytic cells and in cells of the central nervous system
♦
Skeletal manifestations include the Erlenmeyer flask deformity of the femur, osteonecrosis of the femoral head, bone infarcts, and pathologic fractures
MUSCULOSKELETAL NEOPLASMS
Bone Cysts
Simple (Unicameral) Bone Cyst (UBC)
Definition
♦
A benign usually unilocular, intramedullary cyst filled with serous or serosanguineous fluid
Clinical
♦
Most patients are within the first two decades of life. There is a slight male predominance
♦
Patients might be asymptomatic or present with sudden onset of pain from a pathologic fracture
♦
About 80% of cases are seen in either the humerus or the femur. Other relatively common sites include the ilium and the calcaneus (especially in somewhat older patients)
X-Rays and Imaging Findings
♦
Geographic, lytic, and cystic lesions (often metaphyseal)
♦
May encroach the epiphysis in skeletally immature individuals
♦
As the bone lengthens at the physeal end, the cyst may appear to “move” into a diaphyseal location
Macroscopic
♦
The lesion is typically a unilocular intramedullary cyst filled with a clear or straw-colored fluid. In cases that have had a superimposed fracture, the fluid may be bloody or blood-tinged. In cases that are healing, there may be a few septae or loculations within the cyst
Microscopic
♦
Cyst lined by a thin, fibrovascular membrane
♦
Irregular fragments of membranous, fibrovascular tissue
♦
Hemosiderin, granulation tissue, or mild focal chronic inflammatory cells
♦
Pink, cementum-like, rounded material may be seen
These may be examples of the Liesegang phenomenon , forming from diffusion and precipitation of supersaturated solutions
Intraosseous Ganglion
Definition
♦
An intramedullary, mucin-filled, fibrous-lined lesion
Clinical
♦
Wide age range, often with pain, and incidental finding
♦
The distal and proximal tibia , femur, ulna, and the hands and feet are commonly involved
X-Rays and Imaging Findings
♦
Geographic lesions that are epiphyseally located and show sclerotic margins
♦
The joint articular surface is usually normal (as opposed to subchondral osteoarthritic cysts)
Microscopic
♦
Mainly myxoid tissue mixed with fibroblasts
♦
Fibrous tissue may be haphazardly interspersed or arranged in the form of septa
♦
The outer layer is often heavily collagenized
Differential Diagnosis
♦
Extragnathic fibromyxoma
♦
Chondromyxoid fibroma
♦
Subchondral cysts of degenerative joint disease
Aneurysmal Bone Cyst
Definition
♦
A benign cyst composed of blood-filled spaces separated by connective tissue septae containing fibroblasts, osteoclasts, and woven bone. The lesion may be primary (contain no underlying bone tumor) or secondary (arise in the setting of a prior benign or malignant bone tumor)
♦
A solid (noncystic) variant of this lesion has been described (solid aneurysmal bone cyst or giant cell reparative granuloma)
Clinical
♦
There is a wide age range, but it mostly occurs between ages 5–25 years
♦
Pain of a few weeks duration is the most common presenting symptom
♦
Most primary ABCs of long bones are metaphyseal in location
♦
Secondary ABCs follow the site of predilection of their primary lesions
♦
Secondary ABC may represent a change secondary to a variety of different bone neoplasms, including:
Giant cell tumor
Nonossifying fibroma
Giant cell reparative granuloma
Fibrous dysplasia
Chondromyxoid fibroma
Chondroblastoma
Osteoblastoma
Unicameral (simple) bone cyst
Hemangioma
Osteosarcoma
♦
The diagnosis of ABC is essentially that of exclusion
♦
Some of these lesions spontaneously regress
♦
There appears to be molecular evidence that primary ABC (including the solid ABC) are true neoplasms since they have a consistent translocation involving the USP6 gene. This translocation is lacking in secondary ABC
X-Rays and Imaging Findings
♦
A benign lesion that is often multicystic, rapidly expansile, and locally destructive
♦
The ABCs of long bones may be central, eccentric, or less commonly parosteal
♦
In the initial (or incipient) phase, there is a small lytic lesion that does not expand the bone
♦
In the stable phase, the X-rays have a characteristic picture, with expanded bone and a “shell” around the lesion, along with trabeculations coursing within it
♦
In the healing phase, progressive ossification results in a coarsely trabeculated bony mass
♦
Fluid–fluid levels on CT scan or MRI are characteristic (Fig. 21.9A)
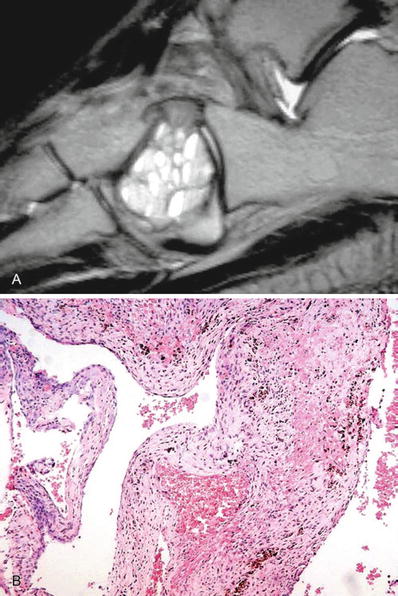
Fig. 21.9.
Aneurysmal bone cyst – MRI showing fluid levels within a well-delineated lesion limited to the cuboid (A). Microscopic examination showed hypocellular fibrous septae with some reactive osteoid and hemosiderin (B).
Macroscopic
♦
Grossly (if intact), the lesions have a thin osseous bony shell surrounding a honeycombed mass with cavernous vascular spaces that “ooze” blood like a sponge
♦
Older cysts may contain serosanguinous fluid rather than blood
♦
A careful search should be made for solid areas that may represent the precursor lesion
Microscopic
♦
Fibrous walls that contain varying proportions of osteoid, chondroid, giant cells, and inflammation (Fig. 21.9B)
♦
Cavernous spaces that are filled with blood
♦
Lack the smooth muscle wall and endothelial cells of blood vessels
♦
The mineralizing component may have a chondroid aura , unusual in any other lesion
♦
The chondroid usually has a fibrillary or chondromyxoid quality and may be focally calcified
♦
Mitotic figures may be numerous, particularly in the areas of osteoid formation
♦
The stromal cells lack anaplasia
♦
The solid variant of ABC (reparative giant cell granuloma) lacks the blood-filled spaces but is otherwise similar to the usual ABC
♦
Primary ABCs is characterized by a recurrent translocation (2, 3) involving the USP6 gene leading to constitutive activation of USP6 (ubiquitin-specific protease) under the control of strong promoters of multiple gene partners (CDH11, OMD, COL1A1, ZNF9, TRAP150)
♦
Up to 70% of primary aneurysmal bone cysts carry translocation involving the USP6 gene
♦
The rearrangement of USP6 in the neoplastic stromal cell population is readily identified using break-apart FISH probes
Differential Diagnosis
♦
Telangiectatic osteosarcoma
Occasionally misleading radiographic appearance
Presence of cellular anaplasia and atypical mitotic figures
Irregular lace-like deposition of osteoid
♦
Ossifying hematoma and pseudotumor of hemophilia
Subperiosteal hematomas
Radiographically, may mimic a parosteal ABC
Hemosiderin, zonation, and organized appearance of bone
♦
Giant cell tumor with ABC component
Older patients
Sheets of giant cells
No marked osteoblastic change
Bone-Forming Lesions
Osteoid Osteoma
Definition
♦
A benign bone-forming tumor characterized by small size, limited growth potential, and often severe pain
Clinical
♦
Majority found within the first three decades
♦
Presentation
Severe, unremitting pain
Relief of pain by aspirin is seen in many patients
X-Rays and Imaging Findings
♦
Classic location: long bones
♦
The lesion has sclerotic borders on X-rays. It is characterized by a central radiolucent nidus and surrounding dense bone, in most cases
♦
The nidus is lucent, but it may have a small central radiodense spot of calcification
♦
Demonstration of the nidus may require tomograms or CT scans
♦
Osteoid osteomas of the joints can be difficult to detect by plain films
♦
Osteoid osteomas are characteristically hot on technetium pyrophosphate bone scans
Macroscopic
♦
The nidus is red, spherical, and gritty
♦
Usually can be “shelled” out from the surrounding bone
Microscopic
♦
A benign neoplasm consisting of a nidus and surrounding reactive, sclerotic bone. There is a sharp demarcation between the nidus and the surrounding bone (Fig. 21.10)
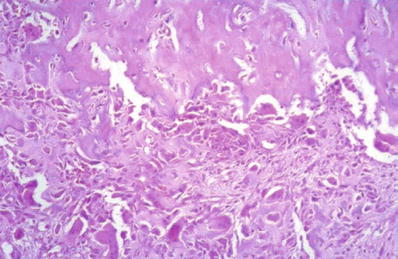
Fig. 21.10.
Osteoid osteoma . There is fibrovascular tissue rich in osteoblasts that is part of the nidus (in the lower part of the picture). It is sharply demarcated from the surrounding dense lamellar bone.
♦
The nidus is a highly vascular, sharply defined osteoblastic proliferation, usually <1.5 cm
♦
The nidus is variably ossified. In some cases, it is poorly ossified with a richly vascularized stroma. In others, it can be heavily ossified, with calcific or lacy osteoid composed of osteoid rimmed with plump osteoblasts
♦
Although mitotic activity can be seen within the nidus, there are no atypical mitotic figures
♦
The woven bone shows prominent osteoblastic rimming
♦
0.1–0.2 cm zone of less trabeculated fibrovascular tissue around the nidus
♦
Sclerotic compact or spongy lamellar bone surrounds the fibrovascular tissue
Differential Diagnosis
♦
Clinical and radiologic mimics
Intracortical abscess (such as Salmonella infection)
Sclerosing osteomyelitis (of Garre)
Enostosis
Aseptic necrosis
Stress fracture
Langerhans cell histiocytosis
Metastasis
♦

Histologic mimics
Get Clinical Tree app for offline access
Osteoblastoma
•
Osteoid osteomas are always <2 cm (most cases are less than 1.5 cm)
•
The previously termed “giant” osteoid osteomas (>2 cm) were probably examples of osteoblastomas

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

