Normal Structure & Function
Blood is an extremely complex fluid, composed of both formed elements (red cells, white cells, platelets) and plasma. Red blood cells (erythrocytes) are the most common formed elements, carrying oxygen to the cells of the body via their main component, hemoglobin. White blood cells are generally present at about 1/700th the number of erythrocytes and function as mediators of immune responses to infection or other stimuli of inflammation. Platelets are the formed elements that participate in coagulation. Plasma is largely water, electrolytes, and plasma proteins. The plasma proteins most important in blood clotting are the coagulation factors. Because blood circulates throughout the body, alterations in normal blood physiology—either formed elements or plasma proteins—may have widespread adverse consequences.
Although the mature formed elements of blood are quite different from each other in both structure and function, all of these cells develop from a common hematopoietic stem cell population, which resides in the bone marrow. The developmental process is called hematopoiesis and represents an enormous metabolic task for the body. More than 100 billion cells are produced every day. This makes the bone marrow one of the most active organs in the body. In adults, most of the active marrow resides in the vertebrae, sternum, and ribs. In children, the marrow is more active in the long bones.
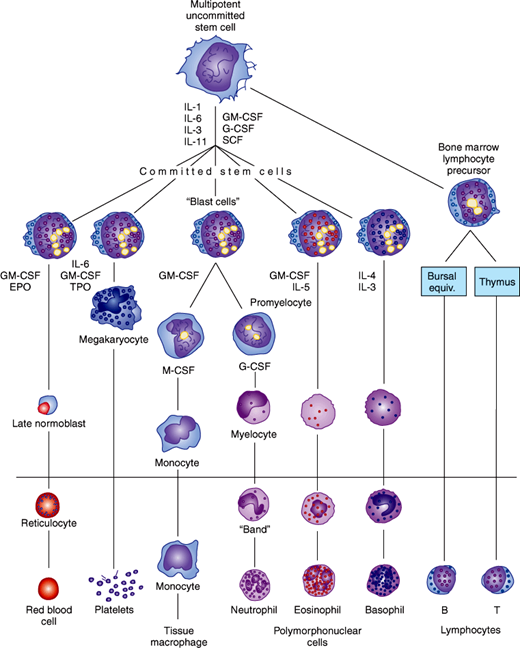
The process of differentiation from stem cell to mature erythrocyte, granulocyte, lymphocyte, monocyte, or platelet is shown in Figure 6–1. It is not clear exactly what early events lead dividing stem cells down a particular path of development, but many different peptides, called cytokines, are clearly involved (Table 6–1); see also Chapter 3. Perhaps because mature white blood cells have a much shorter half-life in the circulation, white blood cell precursors usually outnumber red blood cell precursors by a ratio of 3:1 in the bone marrow.
Figure 6–1
Hematopoiesis: development of the formed elements of blood from bone marrow stem cells. Cells below the horizontal line are found in normal peripheral blood. The principal cytokines that stimulate each cell lineage to differentiate are shown. (EPO, erythropoietin; TPO, thrombopoietin; CSF, colony-stimulating factor; G, granulocyte; M, macrophage; IL, interleukin; SCF, stem cell factor.) See Table 6–1 for details. (Redrawn, with permission, from Ganong WF. Review of Medical Physiology, 22nd ed. McGraw-Hill, 2005.)
| Cytokine | Cell Lines Stimulated | Cytokine Source |
|---|---|---|
| IL-1 | Erythrocyte | Multiple cell types |
| Granulocyte | ||
| Megakaryocyte | ||
| Monocyte | ||
| IL-3 | Erythrocyte | T lymphocytes |
| Granulocyte | ||
| Megakaryocyte | ||
| Monocyte | ||
| IL-4 | Basophil | T lymphocytes |
| IL-5 | Eosinophil | T lymphocytes |
| IL-6 | Erythrocyte | Endothelial cells |
| Granulocyte | Fibroblasts | |
| Megakaryocyte | Macrophages | |
| Monocyte | ||
| IL-11 | Erythrocyte | Fibroblasts |
| Granulocyte | Osteoblasts | |
| Megakaryocyte | ||
| Erythropoietin | Erythrocyte | Kidney |
| Kupffer cells of liver | ||
| SCF | Erythrocyte | Multiple cell types |
| Granulocyte | ||
| Megakaryocyte | ||
| Monocyte | ||
| G-CSF | Granulocyte | Endothelial cells |
| Fibroblasts | ||
| Monocytes | ||
| GM-CSF | Erythrocyte | Endothelial cells |
| Granulocyte | Fibroblasts | |
| Megakaryocyte | Monocytes | |
| T lymphocytes | ||
| M-CSF | Monocyte | Endothelial cells |
| Fibroblasts | ||
| Monocytes | ||
| Thrombopoietin | Megakaryocyte | Liver, kidney |
The major hormone that stimulates the production of erythrocytes (erythropoiesis) is erythropoietin. This peptide is produced by the kidneys and regulates red blood cell production by a feedback system: When blood hemoglobin levels fall (anemia), oxygen delivery to the kidneys falls, and they produce more erythropoietin, causing the marrow to produce more red cells. When hemoglobin levels rise, the kidney produces less erythropoietin and the marrow fewer red cells.
For white blood cells, the situation is more complex. The most common cells are the granulocytes, so named because their cytoplasms are filled with granules. Of these, the neutrophils are the most prevalent and the most important cells in producing inflammation. Granulocyte production (myelopoiesis) can be affected by many cytokines at different stages of development. Figure 6–1 shows that interleukin-3 (IL-3), granulocyte colony-stimulating factor (G-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) are the most important. All three proteins have been purified, sequenced, and cloned. The latter two proteins are used therapeutically. Unlike G-CSF, GM-CSF also stimulates the maturation of a different white blood cell line, the monocyte-macrophage line. These cells are part of the immune system as well (eg, ingesting foreign bacteria) and can reside in skin and other tissues, not just blood. Their function, along with that of the B- and T-lymphocyte populations, is discussed more fully in Chapter 3.
Platelets are not cells but fragments of larger multinucleated cells in the marrow called megakaryocytes. Platelets are crucial to normal blood clotting. Platelet production is also stimulated by multiple cytokines but is dependent mainly on the action of IL-3, IL-6, and IL-11 and thrombopoietin (TPO). This peptide is produced by the liver, kidney, skeletal muscle, and marrow stroma. One model of thrombopoiesis proposes that the production of TPO occurs at a constant rate. However, the amount of this hormone free to interact with platelet precursors rises and falls, probably as a result of uptake by TPO receptors (c-Mpl) on existing platelets in the blood. Therefore, a low platelet count (with a lower mass of c-Mpl) stimulates thrombopoiesis as a result of increased circulating levels of TPO. A second model proposes that low platelet levels can induce increased production of TPO in marrow stromal cells, via various cytokines including platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF). These two models are not necessarily mutually exclusive. Inflammation can also lead to thrombocytosis via IL-6–mediated increases in TPO production by the liver.
For all its complexity and metabolic activity, there is tremendous regulation of the marrow through the interaction of various cytokines. Normally, only the most mature elements in each cell lineage are released into the general circulation, demonstrating this exquisite control over development. Complex negative-feedback mechanisms must be at work to maintain circulating quantities of each formed element at the consistent levels at which they are found.
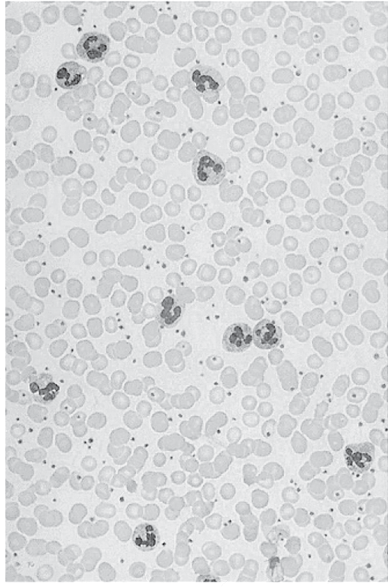
Examination of the appropriateness of blood cell development is best undertaken with the microscope, using the thin blood smear (Figure 6–2). Modern technical equipment, which can optically sort cells by size and various optical reflective parameters, gives important information, especially about whether cell numbers are out of the normal ranges (Table 6–2). However, microscopic examination of the blood smear, usually using Wright stain, gives additional information once an abnormality is detected and should always be done when a blood disorder is suspected on clinical grounds.
Figure 6–2
Normal thin blood smear, seen at low power (40×) with Wright stain. Erythrocytes predominate and can be seen to be thin disks with central pallor (see text). Platelets are the numerous small, dark bodies. Larger cells with lobulated nuclei are mature neutrophils. Lymphocytes and monocytes are not present on this smear.
| Element | Male Adult | Female Adult |
|---|---|---|
| Hemoglobin | 14–18 g/dL | 12–16 g/dL |
| Hematocrit (percentage of blood that is erythrocytes) | 42–50% | 37–47% |
| Red cell count | 4.6–6 × 106/μL | 4.2–5.4 × 106/μL |
| Mean corpuscular volume (MCV) | 80–100 fL | 80–100 fL |
| White blood cell (total) count | 4000–11,000/μL | 4000–11,000/μL |
| Neutrophils | 2500–7500/μL | 2500–7500/μL |
| Lymphocytes | 1500–3500/μL | 1500–3500/μL |
| Monocytes | 200–800/μL | 200–800/μL |
| Eosinophils | 60–600/μL | 60–600/μL |
| Basophils | <100/μL | <100/μL |
| Platelets | 150,000– 400,000/μL | 150,000– 400,000/μL |
Mature red blood cells are biconcave disk-shaped cells filled with hemoglobin, which function as the oxygen-carrying component of the blood. In contrast to most other cells, they do not have nuclei at maturity; their nuclei are extruded during the final phase of erythrocyte development. The presence of erythrocytes with nuclei in the peripheral blood smear suggests an underlying disease state. Normal red cells are about 8 μm in diameter, a size that is larger than the smallest capillaries. However, their biconcave shape gives them enough flexibility to slip through small capillaries and deliver oxygen to the tissues. Once extruded from the bone marrow, individual erythrocytes function for about 120 days before they are removed from the circulation by the spleen.
In a typical blood smear (stained with Wright stain), erythrocytes dominate the microscopic field, and their biconcave disk shape resembles that of a doughnut. There is a thicker outer rim that appears red owing to the hemoglobin present and an area of central pallor where the disk is thinnest. Young erythrocytes (reticulocytes) appear bluer (basophilic) because they still contain some ribosomes and mitochondria for a few days after the nuclei are extruded.
Hemoglobin is the most important substance in the erythrocyte. This protein is actually a tetramer, made of two α-protein subunits and two β-protein subunits (in normal adult hemoglobin, called hemoglobin A). Each α- or β-subunit contains the actual oxygen-binding portion of the complex, heme. Heme is a compound whose centrally important atom is iron; it is this atom that actually binds oxygen in the lungs and subsequently releases it in the tissues of the body. A low level of hemoglobin in the blood, from a variety of causes (see later discussion), is anemia, the most common general blood disorder.
The granulocytes are the most common white blood cells; of these, neutrophils are most abundant, followed by eosinophils and basophils (Table 6–2). Developmentally, all three types are similar: As they mature, their nuclei become more convoluted and multilobed, and each develops a cytoplasm filled with granules. These granules contain a variety of enzymes, prostaglandins, and mediators of inflammation, with specific factors dependent on the cell type. Early progenitor cells for each type of granulocyte (“blasts”) are indistinguishable on microscopic examination of the bone marrow, but under the influence of different cytokines, they become morphologically distinct cell types.
Basophils contain very dark blue or purple granules when stained with either Giemsa or Wright stain. Basophil granules are large and usually obscure the nucleus because of their density. Normally, basophils function in hypersensitivity reactions (as described in Chapter 3). However, their numbers can be increased in diseases not associated with hypersensitivity, such as chronic myelogenous leukemia.
Eosinophils contain large, strikingly “eosinophilic” granules (staining red with Wright or Giemsa stain). Eosinophil nuclei are usually bilobed. Normally, eosinophils function as part of the inflammatory response to parasites too large to be engulfed by individual immune cells. They are also involved in some allergic reactions.
Neutrophils contain granules that are “neutrophilic” (ie, neither eosinophilic nor basophilic). Although they predominate in the blood, their major function is actually in the tissues; they must leave the blood by inserting themselves between the endothelial cells of the vasculature to reach sites of injury or infection. Their granules contain highly active enzymes such as myeloperoxidase, which, along with the free radical oxygen ions produced by membrane enzymes such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, kill bacteria that neutrophils ingest via endocytosis or phagocytosis. They are the “first line of defense” against bacterial pathogens, and low numbers of them (leukopenia) lead directly to a high incidence of significant bacterial infections (see later discussion). Of all the cells produced by the bone marrow, the neutrophils comprise the greatest fraction. Their life span in blood, only 8 hours, is much shorter than that of any other cell type. Evidence of their importance and their short survival is commonly manifested, because examination of the blood smear under the microscope in a patient with an active infection may show not only increased numbers of mature, multilobed neutrophils (neutrophilia) but also increased numbers of less mature cells. These less mature cells, released from a large storage pool in the bone marrow, are called bands and have a characteristic horseshoe-shaped nucleus that is not yet fully lobulated. The phenomenon of finding these cells in the peripheral blood is called a left shift of the granulocyte lineage.
Both monocytes and lymphocytes arise from the common stem cell. It is the widespread pluripotential ability of stem cells to differentiate into these cells in addition to the granulocytes, erythrocytes, and platelets that makes bone marrow transplantation a therapeutic option for immune system disorders and malignancies. Monocytes have a very long life span, probably several months, but spend only about 3 days in the circulation. They mostly reside in tissues and act there as immune cells that engulf (phagocytose) bacteria and subsequently can “present” components of these bacteria to lymphocytes in a way that further amplifies and refines the immune response (Chapter 3). On blood smear evaluation, monocytes are the largest cells seen, with irregular but not multilobed nuclei and pale blue cytoplasm, often with prominent vacuoles.
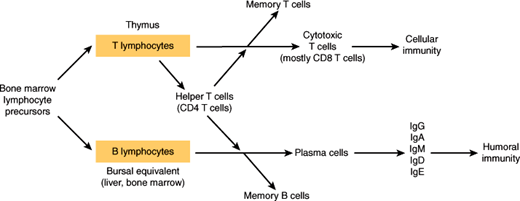
Lymphocyte precursors leave the marrow early and require extramedullary (outside of the marrow) maturation to become normally functioning immune cells in either the blood or the lymphatic system (Figure 6–3). Their crucial roles in recognizing “self” versus “nonself” and in modulating virtually all aspects of the immune response are described in Chapter 3. On microscopic examination of the blood smear, lymphocytes are small cells, slightly larger than an erythrocyte, with dark nuclei essentially filling the entire cell; only a thin rim of light blue cytoplasm is normally seen. Granules are sparse or absent.
Platelets are the smallest formed elements in the blood. They are fragments of larger, multinucleated cells, which are the largest discrete constituents of the bone marrow (megakaryocytes), but platelets have no nuclei of their own. Most platelets remain in the circulation, but a substantial minority is trapped in the spleen; this phenomenon becomes important in a variety of immune-mediated decreases in platelet count (thrombocytopenia; see later discussion). In the setting of a normal platelet count, they have a circulatory half-life of about 10 days. In cases of thrombocytopenia, their half-life decreases, as they are consumed in the routine maintenance of vascular integrity.
Platelets are integral components of the coagulation system. Their membranes provide an important source of phospholipids (PLs), which are required for the function of the coagulation system proteins (Figure 6–4), and contain important receptors that allow attachment to endothelial cells (platelet adhesion) so that a platelet plug can be formed in response to blood vessel injury. This prevents further blood loss after trauma and limits the coagulation response to the site of injury rather than letting coagulation proceed inappropriately.
The cytoplasm is also important for platelet function, particularly the intracellular dense granules and alpha granules. The phenomenon of platelet activation is also called “degranulation” and can be initiated by exposure of platelets to the activated blood coagulation factor thrombin, adenosine 5′-diphosphate (ADP), or collagen. This last reaction is probably the most important, occurring when collagen, normally in the basement membrane below the endothelial cells, is exposed to the blood after injury. Platelet activation can also be induced by exposure to platelet-activating factor (PAF) (a neutrophil-derived phospholipid cytokine), thromboxane A2, serotonin, and epinephrine.
During platelet activation, the dense and alpha granules release further activators of platelet activity, such as ADP, and platelet factor 4, which can also bind to endothelial cells. It is important because it binds to the most commonly used therapeutic anticoagulant, heparin (see later discussion). After activation, platelets change shape from discoid to spherical with filopodial extensions and finally to a flat shape that allows for adequate coverage of the site of vessel injury. The last step in platelet activity is platelet aggregation, where platelets stick to each other, firming up the platelet plug. On examination of the blood smear, platelets are small, irregularly shaped blue or purple granular bodies. In conditions in which platelet numbers are rising as a result of increased marrow activity, more immature platelets can be identified by their larger size.
Checkpoint
What is the ratio of red blood cells to white blood cells in the bloodstream?
What is the number of cells produced daily by the bone marrow?
What are the different formed elements of blood and how can they and their subtypes be distinguished?
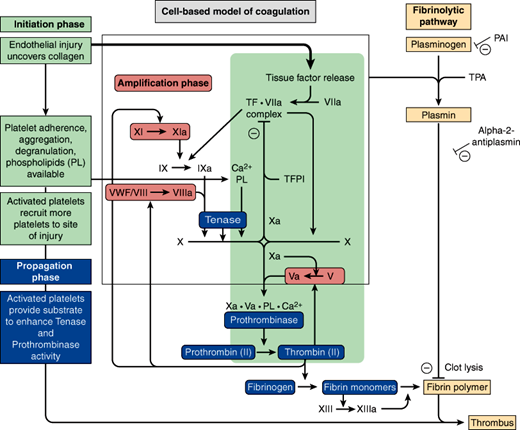
The coagulation system, diagrammed in Figure 6–4, is a highly complex, regulated interaction of cells and plasma proteins. The coagulation system provides for immediate activation when control of bleeding (hemostasis) is required and confines its activity to the site of blood loss. Otherwise, coagulation might occur throughout the entire circulatory system, which would be incompatible with life.
The major components of hemostasis are platelets (discussed previously), endothelial cells (lining the blood vessels), other tissue factor (TF)–bearing cells and the coagulation factors, which are plasma proteins. The end result of the activated coagulation system is the formation of a complex of cross-linked fibrin molecules and platelets that terminate hemorrhage after injury. To maintain a well-regulated balance between prothrombotic and antithrombotic factors, the sophisticated coagulation system provides several points of control (Figure 6–4).
The coagulation factors do not generally circulate in active forms. Most of them are enzymes (serine proteases) and remain dormant until they are needed. This is accomplished by having other enzymes (the other proteases in the coagulation system) available that can cleave the inactive factors into active ones. All of the factors have roman numerals, and the inactive forms are written without annotation (eg, factor II, also known as prothrombin). The activated forms of the factors are signified by the letter “a” (eg, factor IIa, also known as thrombin).
Most of the coagulation factors are made by the liver, but factor XIII derives from platelets and factor VIII is made by endothelial cells. Factors II, VII, IX, and X are particularly important factors (Table 6–3) because they are all dependent on the liver enzyme γ-carboxylase. Gammacarboxylase is dependent on vitamin K, and the oral anticoagulant warfarin acts by interfering with vitamin K activity. Two of the anticoagulant proteins, protein S and protein C (see later discussion), are also vitamin K dependent.
| Name | Production Source |
|---|---|
| Procoagulant factors | |
| Factor I (fibrinogen) | Liver |
| Factor II (prothrombin) | Liver |
| Factor III (tissue thromboplastin) | Tissue |
| Factor IV (calcium) | … |
| Factor V (proaccelerin) | Liver |
| Factor VI (obsolete = factor Va) | … |
| Factor VII (proconvertin) | Liver |
| Factor VIII (antihemophilic factor) | Endothelial cells |
| Factor IX (Christmas factor) | Liver |
| Factor X (Stuart-Prower factor) | Liver |
| Factor XI (plasma thromboplastin antecedent) | Liver |
| Factor XII (Hageman factor) | Liver |
| Factor XIII (fibrin-stabilizing factor) | Platelets |
| Anticoagulant factors | |
| Antithrombin | Liver |
| Protein C | Liver |
| Protein S | Liver |
| Plasminogen | Liver |
| Tissue factor pathway inhibitor | Endothelial cells |
Hemostasis is divided into three major processes: primary hemostasis, secondary hemostasis, and fibrinolysis.
Primary hemostasis involves vasoconstriction and platelet adhesion and activation at sites of endothelial injury. Collagen and thrombin activate platelets, leading to an increase in intracellular calcium, secretion of platelet granules, and activation of various signaling pathways.
Secondary hemostasis is the process whereby fibrin is formed. The classical coagulation cascade, involving the intrinsic, extrinsic, and common pathways, better describes coagulation in vitro, as tested by the coagulation assays: activated partial thromboplastin time (aPTT) and prothrombin time (PT). The cell-based model of coagulation has replaced the coagulation cascade as a more accurate depiction of the in vivo coagulation process (Figure 6–4). Secondary hemostasis is further divided into three overlapping phases: initiation, amplification, and propagation.
Initiation occurs at the surface of injured cells. It starts with the release of TF by the injured cells. TF, also called thromboplastin, is a lipid-rich protein material that is exposed to plasma upon injury to the vascular wall. It directly activates factor VII, forming the complex TF-VIIa, which activates both factor IX and factor X. Together factor Xa (an enzyme) and Va (a cofactor, activated from factor V by Factor Xa) on the surface of the injured cell catalyze the conversion of prothrombin (II) to thrombin (IIa). Thrombin, a serine protease, cleaves the ubiquitous plasma protein fibrinogen into fibrin monomers, small insoluble proteins that can polymerize with each other to form the complex fibrin; however, the amount of thrombin formed at the site of the injured cell is insufficient by itself to produce enough fibrin to stabilize the platelet plug.
Amplification, unlike the initiation phase, occurs at the surface of platelets. During this phase, thrombin produced in the initiation phase activates platelets and coagulation factors V, VIII, and XI found on the platelet surface. Factor VIII is normally complexed to von Willebrand factor (vWF), the protein that allows platelets to adhere to endothelial cells. Thrombin activates Factor VIII by releasing it from vWF. It also activates both V and XI, which allows them to bind to the platelet surface. Factor XIa then catalyzes the activation of IX to IXa, providing supplemental factor IXa at the platelet surface.
Propagation involves activated platelets recruiting other circulating platelets to the site of vessel injury and the formation of two major complexes: tenase and prothrombinase, which are crucial to fibrin production. Factors VIIIa and IXa form the tenase complex on the surface of platelets in the presence of PLs and calcium (VIIIa-IXa-Ca2+-PL). Together, they activate factor X on the platelet surface. Factor Xa then forms the prothrombinase complex with factor Va on the platelet surface, again in the presence of PLs and calcium (Xa-Va-Ca2+-PL). This complex catalyzes the cleavage of prothrombin (II) to thrombin (IIa) and can convert multiple molecules per complex. As the activated platelets recruit more circulating platelets to the site of injury, a critical mass of platelets leads to a surge of thrombin generation. This, in turn, leads to enough fibrin formation to stabilize the platelet plug. This fibrin polymer is further solidified by chemical cross-links catalyzed by factor XIIIa, which itself is activated by thrombin. Factor XIIIa also incorporates α2-antiplasmin into the clot to protect it from fibrinolytic proteases.
Fibrinolysis involves the process of breaking down fibrin into its degradation products. Plasmin is the main catalytic enzyme in this process. It is a serum protease that cleaves fibrin, resulting in breakup of the clot and creating fibrin degradation products that inhibit thrombin. Thrombin, working in a negative feedback fashion, actually helps catalyze the formation of plasmin from the inactive precursor protein, plasminogen. Plasminogen can also be cleaved by tissue plasminogen activator (t-PA) to form plasmin; t-PA and related proteins are used clinically to break up clots that form in coronary arteries in patients with a new myocardial infarction, as well as in cerebral arteries in patients with a new stroke. Inhibitors of fibrinolysis include plasminogen activator inhibitor and α2-antiplasmin.
In addition to the fibrinolytic pathway, checks on the coagulation system (ie, the anticoagulant system) also involve various feedback loops and inhibitors. Factor Xa binds to another plasma (and lipid-bound) protein called tissue factor pathway inhibitor (TFPI). TFPI not only inhibits further activity of factor Xa itself but also prevents Xa from binding to the platelet surface, and the combination of factor Xa and TFPI greatly inhibits TF-VIIa complex. Furthermore, downstream prothrombinase activity can be sustained only if the initial injury continues to generate enough factor IXa and VIIIa (in the form of the tenase complex) to activate more factor X on platelet surfaces.
Other anticoagulants include a group of inhibitors of the coagulation factors. They are composed of antithrombin (AT), protein S, and protein C (see later discussion). AT is a protease inhibitor and physically blocks the action of the serine proteases in the coagulation system. Its activity is enhanced up to 2000-fold by heparin. Protein C, activated by thrombin, cleaves factor Va into an inactive form so that the prothrombinase complex cannot cleave prothrombin (II) into thrombin. Protein C requires protein S as a cofactor. This complex also inactivates factor VIIIa.
Assays are available for determining both the absolute level and the activity of each of the coagulation factors, but in practice there are two common in vitro tests of coagulation function, both reported as “seconds required to form a clot”: the PT and the aPTT. The tests are designed in such a way that the results will be prolonged out of the normal range in different pathologic states, but significant alterations in the coagulation pathway inevitably lead to changes in both tests because of the multiple interactions of the involved factors.
PT assesses the “extrinsic” TF-dependent and common pathways of the classical coagulation cascade and is used clinically to monitor the effects of warfarin. Because all vitamin K–dependent factor levels are lowered by warfarin, eventually the aPTT will also become abnormal with high enough doses; but factor VII has the shortest half-life of those factors, so its levels fall first. Because of its critical role in clotting, thrombin is the principal factor whose activity must be reduced to achieve and maintain therapeutic anticoagulation.
The aPTT assesses the “intrinsic” non–TF-dependent and common pathways and is prolonged most easily when there are reduced levels of factor VIII or factor IX activity, regardless of whether these factors are present at low concentrations or are present at normal concentrations but are being actively inhibited by other molecules. The aPTT is also very sensitive to the presence of heparin bound to AT and is used to monitor the anticoagulant effects of unfractionated heparin. Low-molecular-weight heparins (a specific purified subset of unfractionated heparin) in combination with AT preferentially inhibit factor Xa. In the doses of low-molecular-weight heparins usually given for prevention or treatment of thrombosis, the aPTT will not be prolonged (at least not into the usual “therapeutic range” for unfractionated heparin) despite good evidence of anticoagulation efficacy if factor Xa activity is measured directly.
Checkpoint
Name the vitamin K–dependent clotting factors and the organ in which they are synthesized.
What are the two major complexes found on the surface of activated platelets that are important to the surge in thrombin production? Describe the coagulation factors that form these complexes and what the complexes specifically activate.
What is the main catalytic enzyme in fibrinolysis? Name two inhibitors of fibrinolysis.
Overview of Blood Disorders
Disorders of red cells, white cells, and platelets are separated for discussion because one or the other is found to be the most abnormal during laboratory testing. However, because of the clonal nature of hematopoiesis, many disorders affect all the formed elements of the blood. This is perhaps best demonstrated in the “blast crisis” phase of chronic myelogenous leukemia, in which the majority of both myeloid and lymphoid cells in the blood may be shown to express an identical gene rearrangement, called bcr-abl or Philadelphia chromosome, that has arisen in a single abnormal progenitor cell.
There are many red cell abnormalities, but the principal ones are a variety of anemias. Anemia is defined as an abnormally low hemoglobin concentration in the blood. There are several methods of classification, but the prevailing systems are based on red cell size and shape.
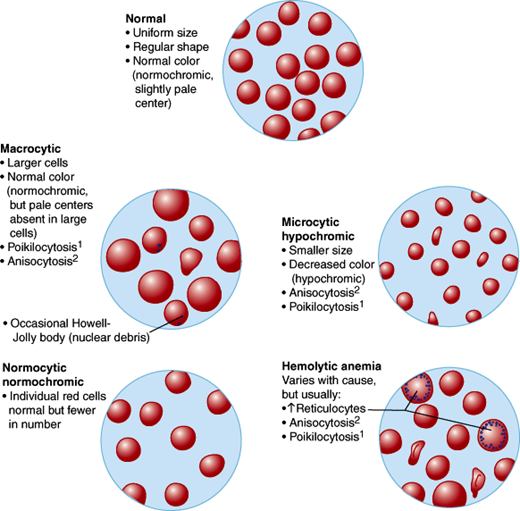
In normal persons, erythrocytes are of uniform size and shape, and the automated blood count shows a mean corpuscular volume (MCV) near 90 fL, which is the estimated volume of a single cell. Automated systems usually report abnormalities of red cells as changes in hemoglobin concentration, red cell number, and MCV. Small cells (with low MCVs) are termed microcytic, and cells larger than normal are termed macrocytic. The relative nonuniformity of cell shapes (poikilocytosis) or sizes (anisocytosis) can further aid in subclassifying erythrocyte disorders.
The morphologic classification of anemias is set forth in Table 6–4 and Figure 6–5. In general, the microcytic anemias are due to abnormalities in hemoglobin production, either in number of hemoglobin molecules per cell or in type of hemoglobin molecules (hemoglobinopathies). Iron deficiency anemia resulting from chronic blood loss and the thalassemias are examples of microcytic anemia.
Figure 6–5
Thin blood smear appearance of erythrocytes in the different morphologic types of anemias. (1, Poikilocytosis [variation in shape]; 2, anisocytosis [variation in size].) (Redrawn, with permission, from Chandrasoma P et al. Concise Pathology, 3rd ed. Originally published by Appleton & Lange. Copyright © 1998 by The McGraw-Hill Companies, Inc.)
| Type | MCV | Common Causes |
|---|---|---|
| Macrocytic | Increased | Folic acid deficiency |
| Vitamin B12 deficiency | ||
| Liver disease | ||
| Alcohol | ||
| Hypothyroidism | ||
| Drugs (sulfonamides, zidovudine, antineoplastic agents) | ||
| Myelodysplastic syndromes | ||
| Microcytic | Decreased | Iron deficiency |
| Thalassemias | ||
| Normocytic | Normal | Aplastic anemia |
| Anemia of chronic disease | ||
| Chronic kidney disease | ||
| Hemolytic anemia | ||
| Spherocytosis |
The macrocytic anemias reflect either abnormal nuclear maturation or a higher fraction of young, large red cells (reticulocytes). When the nuclei of maturing red cells appear too young and large for the amount of hemoglobin in the cytoplasm, the macrocytic anemia is termed megaloblastic. These anemias are most often due either to vitamin deficiencies (vitamin B12 or folic acid) or drugs that interfere with DNA synthesis. Abnormal nuclear maturation can also be due to clonal proliferation in the bone marrow, producing preleukemic states termed the myelodysplastic syndromes.
The normocytic anemias can be due to multiple causes: decreased numbers of red cell precursors in the marrow (primary failure called aplastic anemia, replacement of marrow elements with cancer, certain viral infections, or autoimmune inhibition called pure red cell aplasia), low levels of erythropoietin (resulting from chronic kidney disease), or chronic inflammatory diseases that affect the availability of iron in the marrow. Other normocytic anemias can be secondary to decreased life span of the cells that are produced. Examples of this phenomenon are acute blood loss; autoimmune hemolytic anemias, in which antibodies or complement bind to red cells and cause their destruction; sickle cell anemia, in which the abnormal hemoglobin polymerizes and obliterates the usual resilience of the red cell; and hereditary spherocytosis or hereditary elliptocytosis, in which defects in the erythrocyte membrane affect their ability to squeeze through the capillary microcirculation.
Anemias are very common. In contrast, an elevated hemoglobin concentration, termed erythrocytosis, is uncommon. Elevations in hemoglobin concentration can occur as a secondary phenomenon because of increased erythropoietin levels, such as that found in smokers or people who live at high altitudes (whose low blood oxygen levels stimulate erythropoietin production). Some tumors, especially renal tumors, can also make erythropoietin. Primary polycythemia is an abnormality of the bone marrow itself. This myeloproliferative syndrome leads to an increased red cell mass and consequent low erythropoietin levels by the negative-feedback mechanism discussed previously.
Abnormalities in white cell numbers occur commonly (Table 6–5), whereas abnormalities of function are rare. Neoplastic transformation in the form of leukemia (granulocytes and monocytes) or lymphoma (lymphocytes) is fairly common. The leukemias are discussed in Chapter 5.
| Neutrophilia | Neutropenia |
|---|---|
| Increased marrow activity | Decreased marrow activity |
|
|
| |
|
|
Changes in neutrophil count are the most common white cell abnormality detected on the automated blood count. Increased numbers of neutrophils (leukocytosis) suggest acute or chronic infection or inflammation but can be a sign of many conditions. These include stress, because adrenal corticosteroids cause demargination of neutrophils from blood vessel walls.
Decreased numbers of neutrophils (neutropenia) can be seen in overwhelming infection and benign diseases such as cyclic neutropenia (see later discussion) but can also be seen when the bone marrow is infiltrated with tumor or involved by the myelodysplastic syndromes. Many drugs can also directly suppress marrow production, and because neutrophils have the shortest half-life in the blood of any cell produced by the marrow, their numbers may fall quickly.
Lymphocyte numbers can vary substantially (Table 6–6). Lymphocyte counts are classically elevated in viral infections, such as infectious mononucleosis. However, persistent elevations suggest malignancies, particularly chronic lymphocytic leukemia, which may not cause any symptoms and be incidentally discovered on a routine blood count.
|
|
|
|
Decreased lymphocyte counts (lymphopenia) are a common complication of corticosteroid therapy but are most worrisome for immunodeficiency states; HIV directly infects lymphocytes, and the likelihood of opportunistic infections increases as lymphocyte counts fall, resulting in AIDS.
Abnormalities in platelet number are fairly common, particularly low counts (thrombocytopenia). Causes are listed in Table 6–7. Decreased production of platelets occurs when the marrow is affected by a variety of diseases or when TPO production by the liver is impaired, as in cirrhosis. Increased destruction of platelets is much more prevalent. There are three general mechanisms. Because a significant number of platelets normally reside in the spleen, any increase in spleen size or activity (hypersplenism) leads to lower platelet counts. Platelet consumption that is due to ongoing clotting will also lower counts. Most commonly, however, there is immune-mediated consumption caused by either drugs or autoantibodies. The latter are usually directed against the platelet membrane antigen gpIIb/IIIa.
|
|
|
|
|
|
|
|
|
|
|
|
Functional platelet disorders are common, especially the acquired disorders resulting from uremia (renal failure) or aspirin, which inhibits the platelet enzyme cyclooxygenase and decreases platelet aggregability. Inherited abnormalities are unusual with the exception of von Willebrand disease, which results from either quantitative or qualitative defect of vWF, the carrier protein for factor VIII. vWF also acts as a bridge between platelets and the endothelium and thus is crucial for formation of the platelet plug in the coagulation cascade.
Elevations in the platelet count above normal (thrombocytosis) are relatively common and are especially apt to occur in recovery from iron deficiency anemia upon iron repletion. In the myeloproliferative disorders, such as polycythemia, platelet counts are often high. In essential thrombocythemia, platelet counts may be higher than 1,000,000/μL.
The most important coagulation factor disorders are quantitative rather than qualitative and usually hereditary rather than acquired (Table 6–8). Exceptions to this rule are acquired factor inhibitors, which are antibodies that bind to one of the coagulation factors, most often factor VIII. These may or may not cause clinical bleeding problems, but they can be extremely difficult to treat. The quantitative disorders that most commonly cause bleeding are hemophilia A (deficiency of factor VIII) and hemophilia B
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


