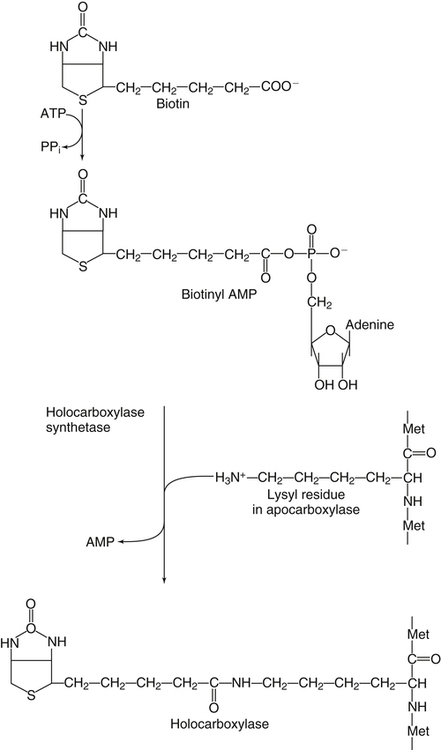
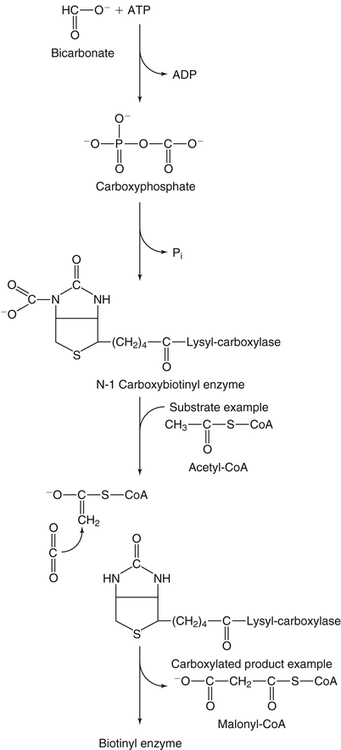
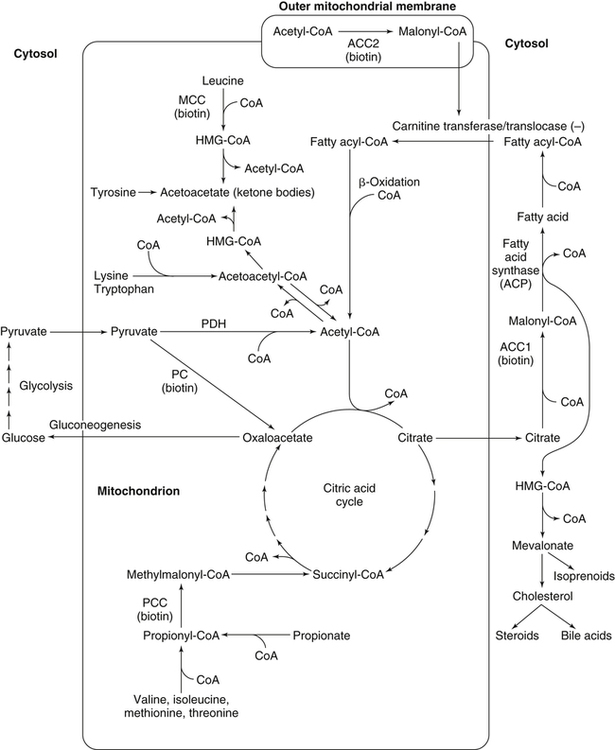
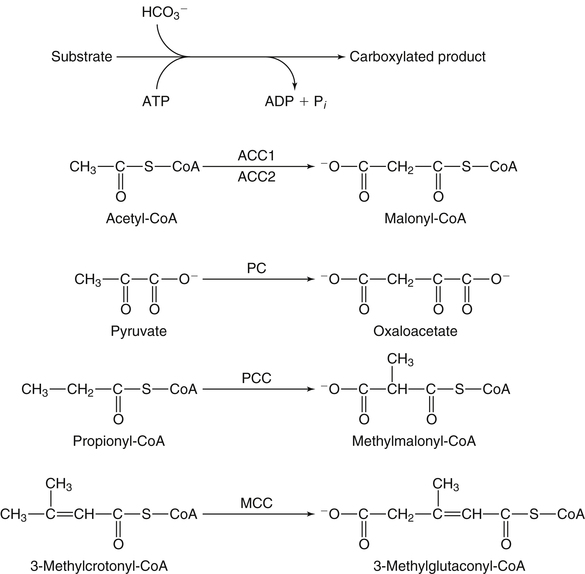
Donald M. Mock, MD, PhD and Nell I. Matthews, BA∗ Biotin is a water-soluble vitamin and is generally classified in the vitamin B complex group. Biotin was discovered by accident in nutritional experiments investigating the protein requirements of rats. Boas (1927) observed that rats fed dried egg white developed scaly dermatitis, hair loss, and neurological signs and pursued this observation by demonstrating that a water-soluble factor in many foodstuffs is capable of curing these signs of deficiency. Egg white contains avidin, a glycoprotein that binds biotin very specifically and tightly. From an evolutionary standpoint, avidin probably serves as a bacteriostat in egg white. Consistent with this possibility is the observation that avidin is resistant to a broad range of bacterial proteases in both the free and the biotin-bound form. Because avidin is also resistant to pancreatic proteases, dietary avidin binds to dietary biotin (and probably any biotin from intestinal microbes) and prevents its absorption. Cooking denatures avidin, rendering this protein susceptible to digestion and unable to interfere with the absorption of biotin. Biotin contains two five-member rings made from a ureido group attached to a tetrahydrothiophene ring (Figure 26-1). A valeric acid side chain is attached to the tetrahydrothiophene ring. The pathway for biosynthesis of biotin was largely elaborated by M.A. Eisenberg and associates working with Escherichia coli (Eisenberg, 1973). Biotin is attached to the inactive apocarboxylase protein by a condensation reaction catalyzed by holocarboxylase synthetase (HCS) (see Figure 26-1). An amide bond is formed between the carboxyl group of the valeric acid side chain of biotin and the ε-amino group of a specific lysine residue in each of the apocarboxylases. These regions of each apocarboxylase contain sequences of amino acids (e.g., Met-Lys-Met at the attachment site) that are highly conserved for the individual carboxylases, and both the N- and C-terminus in HCS are important for recognition of the apocarboxylases (Hassan et al., 2009). In the carboxylase reaction, the carboxyl moiety is first attached to the biotin cofactor (of the holocarboxylase enzyme) at the ureido nitrogen opposite the side chain (Figure 26-2). Dehydration of HCO3− (bicarbonate) to CO2, forming carboxyphosphate, is driven by the hydrolysis of ATP to ADP (see Figure 26-2). The carboxyphosphate then carboxylates a specific nitrogen (N1) in the ureido containing ring of biotin to form N1 carboxybiotinyl-enzyme, and inorganic phosphate is released. These two steps allow HCO3−, which is present at a higher concentration in the cell fluid than is CO2, to be used for formation of the bound, chemically reactive form of CO2. The carboxyl group can then be transferred to a substrate yielding a carboxylated product. Because the valeric acid side chain of biotin is coupled to the side chain of lysine in each holocarboxylase, this CO2 is at the end of a long, flexible chain, allowing the biotinyl coenzyme to be carboxylated at one site and used as a CO2 donor at a second site. The five biotin-dependent mammalian carboxylases are acetyl-CoA carboxylase isoforms 1 and 2 (formerly known as ACCα and ACCβ), pyruvate carboxylase, methylcrotonyl-CoA carboxylase, and propionyl-CoA carboxylase. An overview of the roles of these biotin-dependent carboxylases in the cell is given in Figure 26-3. Acetyl-CoA carboxylase (ACC) catalyzes the incorporation of bicarbonate into acetyl-CoA to form malonyl-CoA (Figure 26-4) and exists in two isoforms. Isoform 1 of ACC (ACC1) is encoded by the ACACA gene and is located in the cytosol. The malonyl-CoA produced by ACC1 is rate-limiting in fatty acid synthesis (elongation). Isoform 2 of ACC (ACC2) is encoded by the ACACB gene and is located on the outer mitochondrial membrane. ACC2 controls fatty acid oxidation in mitochondria through the inhibition of carnitine palmitoyltransferase I by its product malonyl-CoA. Carnitine palmitoyltransferase I catalyzes the rate-limiting step in fatty acid uptake into mitochondria and thus regulates the availability of fatty acids for oxidation. Given that acetyl-CoA is the central compound of intermediary metabolism, it is not surprising that the regulation of ACC1 and ACC2 is complex. The active form of cytosolic ACC1 exists as a very large polymer with a molecular mass in the millions of daltons and is inactivated by dissociation into its protomer units. Citrate activates ACC1 by increasing polymerization. CoA itself activates ACC1 by lowering the Km for acetyl-CoA. ACC1 is inhibited by the products of fatty acid synthesis, the long-chain acyl-CoAs, which also act to depolymerize the enzyme. In addition, ACC1 activity is regulated by covalent modification (phosphorylation) in response to the hormones insulin and glucagon. A high insulin-to-glucagon ratio typical of the immediate postprandial state (fed state with increased blood glucose level) favors dephosphorylation of ACC1 to an active form, whereas a low insulin-to-glucagon ratio (typical of a fasting state) favors phosphorylation to the inactive form. The amount of ACC1 protein also responds to changes in dietary and hormonal conditions. Regulation of ACC2 has not been as extensively studied but appears to operate through inactivation of ACC2 by phosphorylation catalyzed by AMP-activated protein kinase (Cho et al., 2009). The three remaining carboxylases are mitochondrial. Pyruvate carboxylase (PC) is encoded by the PC gene and catalyzes the incorporation of bicarbonate into pyruvate to form oxaloacetate, a central intermediate in carbohydrate, lipid, and amino acid metabolism (see Figure 26-4). The formation of oxaloacetate by PC serves an anaplerotic reaction, replenishing intermediates in the citric acid cycle. This supports synthesis of other intermediates such as α-ketoglutarate, a precursor of glutamate, and citrate, a precursor of cytosolic acetyl-CoA for fatty acid and cholesterol synthesis. Lack of sufficient oxaloacetate for citrate formation limits acetyl-CoA oxidation by the citric acid cycle, thus promoting formation of ketone bodies from acetyl-CoA in the liver. Oxaloacetate is also necessary for aspartate formation and the shuttling of amino acid nitrogen into the urea cycle. The PC reaction also plays a critical role in gluconeogenesis, allowing pyruvate to be converted back to phosphoenolpyruvate via intermediate formation of oxaloacetate. Deficiency of PC is probably the cause of lactic acidemia, central nervous system lactic acidosis, and abnormalities in glucose regulation which are commonly observed in biotin deficiency, genetic biotinidase deficiency, and holocarboxylase synthetase deficiency. Biotinidase is a biotin-amide hydrolase that hydrolyzes biocytin to biotin and lysine, playing a critical role in biotin recycling (see later). PC also plays a role in the formation of the protective myelin sheath that surrounds the axons of certain nerve cells and in the production of neurotransmitters, both of which may contribute to the phenotype of inherited PC deficiency. Propionyl-CoA carboxylase (PCC) catalyzes the incorporation of bicarbonate into propionyl-CoA to form methylmalonyl CoA (see Figure 26-4). Methylmalonyl-CoA then undergoes isomerization to succinyl-CoA, which enters the citric acid cycle; this isomerization is catalyzed by the vitamin B12–dependent enzyme methylmalonyl-CoA mutase. Methylcrotonyl-CoA carboxylase (MCC) catalyzes an essential step in the degradation of the branched-chain amino acid leucine (see Figure 26-4). MCC is composed of two nonidentical subunits: a biotinylated α subunit encoded by the gene MCCC1 and a nonbiotinylated β subunit encoded by the gene MCCC2. MCC is not regulated by small molecules or by dietary or hormonal factors. Deficient activity of MCC leads to metabolism of 3-methylcrotonyl-CoA to 3-hydroxyisovaleric acid, 3-hydroxyisovalerylcarnitine, and 3-methylcrotonylglycine by an alternate pathway (Stratton et al., 2010). Thus increased urinary excretion of these abnormal metabolites reflects deficient activity of MCC. The core histones are designated H2A, H2B, H3, and H4; each nucleosome contains one H3/H3/H4/H4 tetramer and two H2A/H2B dimers. Mammals also express a fifth class of histones: histone H1, which serves as a linker between nucleosomes. Each core histone has a globular domain and a flexible N-terminal tail. The N-terminal tails of core histones protrude from the nucleosomal surface, and covalent modifications of these tails play critical roles in gene regulation (Camporeale et al., 2004; Chew et al., 2006; Kobza et al., 2008). In 1995 Hymes and Wolf discovered that biotinidase can act as a biotinyl-transferase; biocytin serves as the source of biotin, and histones are specifically biotinylated (Hymes et al., 1995). Zempleni and co-workers reported that the abundance of biotinylated histones varies with the cell cycle and that biotinylated histones are increased approximately twofold in activated, dividing lymphocytes compared to quiescent lymphocytes (Stanley et al., 2002). These observations suggested that biotinylation of histones might play a role in regulating DNA transcription as an additional element in the “histone code.” Based on this initial work, it was believed that biotinylation of histones was catalyzed by biotinidase (Hymes et al., 1995). Indeed, approximately 25% of total cellular biotinidase activity is located in the nucleus, and histones are biotinylated enzymatically in a process that is catalyzed in vitro by biotinidase. However, subsequent studies provided evidence that HCS is substantially more important than biotinidase for biotinylation of histones in vivo (Chew et al., 2008; Gralla et al., 2008; Kobza et al., 2008). Fibroblasts from patients with inherited deficiency of HCS exhibit decreased biotinylation of histones. In contrast, biotin can be removed from histones by biotinidase, and debiotinylation of histones is decreased in samples from biotinidase-deficient patients. Current understanding is that HCS plays the predominant role in histone biotinylation and biotinidase plays the predominant role in histone debiotinylation. Biotinylation of distinct lysine residues as a covalent modification of histones that affects DNA transcription has several supporting lines of investigation. Currently, about a dozen biotinylation sites have been identified in histones H2A, H3, and H4. HCS is present in both nuclear and cytoplasmic compartments; in the nucleus, HCS is associated with chromatin. Nevertheless, controversy exists as to whether biotin attachment to histones occurs in vivo (Healy et al., 2009). Although the mechanisms remain to be elucidated, biotin status clearly affects gene expression. Cell culture studies suggest that cell proliferation generates an increased demand for biotin, perhaps stemming from increased synthesis of biotin-dependent carboxylases. Evidence is emerging that this demand is met by an upregulation of biotin transporter expression that is mediated by biotinylation of lysine 12 in histone H4 (H4K12bio) (Gralla et al., 2008). Another function of H4K12bio appears to be chromosomal stability (Camporeale et al., 2007; Chew et al., 2008; Gralla et al., 2008; Wijeratne et al., 2010). Low levels of histone biotinylation have been reported in biotin-deficient cells and model organisms (Chew et al., 2008); reduced histone biotinylation has been linked to increased frequency of retrotransposition events, consistent with a role for histone biotinylation in chromosomal stability. Other roles for biotin in affecting gene expression have been observed. Pioneering studies by Dakshinamurti and co-workers reported a role for biotin in the regulation of the glucokinase gene (Dakshinamurti and Cheah-Tan, 1968). Solorzano-Vargas and colleagues (2002) reported that biotin deficiency reduces messenger RNA (mRNA) levels of HCS, ACC1, and PCC and postulated that a cyclic GMP–dependent signaling pathway is involved in the pathogenesis. Zempleni and co-workers have demonstrated involvement of nitric oxide in the cyclic GMP signaling pathway (Rodriguez-Melendez and Zempleni, 2009; Zempleni et al., 2009). At physiological pH, the carboxyl group of biotin is negatively charged. Thus biotin is at least modestly water-soluble and requires a transporter to cross cell membranes such as enterocytes for absorption (reviewed by Said, 2008). In intact intestinal preparations such as loops and everted gut sacks, biotin transport exhibits two components. One component is saturable at a Km of approximately 10 μM biotin. The other is not saturable, even at very large concentrations of biotin, which is consistent with passive diffusion. Absorption of biocytin is inefficient relative to biotin, suggesting that biotinidase plays a critical role in generating free biotin from biocytin (and dietary protein) and making biotin available for absorption. Human intestinal epithelial cells are highly specialized. Biotin transport must occur across two structurally and functionally different membrane domains: the brush border membrane that faces the intestinal lumen and the basolateral membrane that faces the interstitium, which is in contact with the blood that perfuses the intestine (Said, 2008). In the brush border membrane, transport occurs via a Na+-dependent, electroneutral, carrier-mediated mechanism that saturates at the micromolar range, accounting for the overall limitation in nondiffusion transport (Said, 2008). In the presence of a Na+ gradient, biotin transport occurs against a concentration gradient. This biotin transporter can also transport pantothenic acid and lipoic acid and hence has been named SMVT (sodium-dependent multivitamin transporter). Human SMVT is the product of the SLC5A6 gene, and is exclusively targeted to the apical (brush border) membrane. Biotin transport across the basolateral membrane also occurs by a carrier-mediated mechanism. However, this carrier is Na+-independent, electrogenic, and cannot accumulate biotin against a concentration gradient (Said, 2008). Intestinal biotin transport is upregulated in response to biotin deficiency in both humans and animal models. The mechanism likely involves induction of SMVT mRNA synthesis and an increased number of SMVT transporters per cell. The increase in SMVT is likely mediated by an induction in the activity of P1, which is one of two promoter regions upstream from the SMVT gene (Said, 2008). SMVT is widely expressed in human tissues. Studies by Said and co-workers (Said, 2008) using RNA interference specific for SMVT provide strong evidence that biotin uptake by the liver (and likely many other somatic tissues) occurs via SMVT. Metabolic trapping (e.g., biotin bound covalently to intracellular proteins) is also important. After entering the hepatocyte, biotin diffuses into the mitochondria via a pH-dependent process suggesting that biotin enters the mitochondria in the neutral, protonated form and dissociates into the anionic form in the more alkaline mitochondrial environment, thereby becoming trapped by the charge. The pH of the mitochondrial matrix is about 7.5 compared to about 7.2 for cytosol. Based on a study in rats, biliary excretion of biotin is quantitatively negligible.
Biotin and Pantothenic Acid
Biotin
Biotin Synthesis
Holocarboxylases and Holocarboxylase Synthetase
Biotin-Containing Carboxylases
Carboxylase Mechanism
Acetyl-Coa Carboxylase
Pyruvate Carboxylase
Propionyl-Coa Carboxylase
Methylcrotonyl-Coa Carboxylase
Biotin and Gene Regulation
Biotinylation of Histones
Biotin Digestion and Absorption
Biotin Uptake By Liver
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


