Fig. 16.1
Blood concentration (log-scale) versus time curve of an orally administered medicine
It is the major objective of any medicinal product to yield blood levels of the active substance within the therapeutic window for the period during which the therapeutic effect is desired. At the same time the Cmax should not exceed the MTC and the medicine administration frequency should remain reasonable, preferably not more than two to three times a day. However, this objective is often not easily reached, since the degree of absorption, the absorption and elimination rates, the therapeutic window as well as the intrinsic pharmacological and toxicological activities of an active substance are varying and interactions may occur.
For example, an active substance with a longer elimination half-life may have the advantage of a lower dosing frequency. However, due to the lower elimination rate significant amounts of active substance may still be present in the body when the next dose is administered. Therefore the prescribed dose must be low and it may take several subsequent administrations before the pharmacokinetic equilibrium (the ‘steady state’) is reached and both peak and trough concentration levels will be within the therapeutic window. For medicines that are administered once daily this may even take several days and a booster dose may be needed to obtain a faster therapeutic effect. Active substances possessing a narrow therapeutic window often require therapeutic drug monitoring (TDM). Alternatively, their rate of absorption may need to be regulated through the use of technologically advanced controlled release products. If an active substance has to be administered frequently, because of fast elimination, the use of a slow release product may be considered. This technology may also be considered when toxic effects occur due to peak concentration levels above the MTC.
Concentration versus time relationships (Fig. 16.1) can be translated into concentration versus effect relationships, both for desired and for adverse drug effects (Fig. 16.2). Within the therapeutic window, a low drug blood concentration will generally lead to a low efficacy and increasing the blood concentration will increase efficacy. Moreover, it is considered advantageous when the maximum therapeutic effect is reached already at a concentration that is significantly below the MTC since this would yield a broad safety margin for the patient. This is reflected in an increased margin between the therapeutic and the adverse effect in the curve.
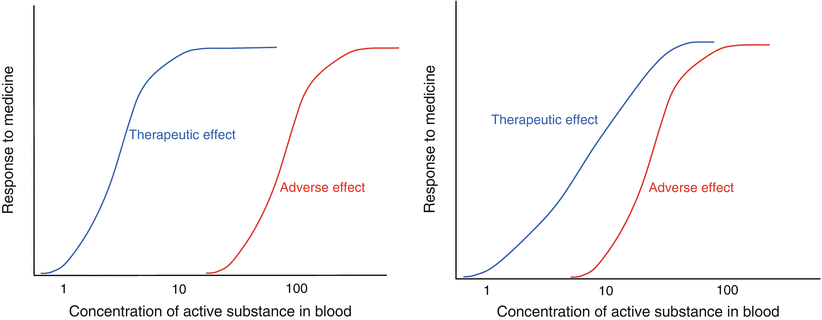
Fig. 16.2
Concentration versus effect relationship of different active substances. On the left hand side an active substance with a steep dose response and a large therapeutic window is shown and on the right hand side an active with a less steep response curve and a smaller safety margin
Whether or not a concentration-effect curve is desired to be steep or flat depends on the drug action and on the intended therapy. For example, for an antihypertensive medicine it may be desirable to tune the effect and a somewhat flatter profile of the curve is preferred. For other situations, such as anti-migraine therapy or infections, tuning the effect is not or less relevant. A steeper curve may have the advantage that the desired effect will be obtained sooner, provided that the maximum effect is reached at concentrations considerably below the MTC.
The impact of variations in the bioavailability on the efficacy and safety of an active substance can be easily understood from these figures. The effect of individual variations in drug sensitivity in relation to variations in drug absorption can also be demonstrated. Examples are given in the box.
Effect of variations in bioavailability on drug efficacy and safety and the relevance of variations in drug sensitivity.
Figure 16.3 (left panel) shows the effects of an increased bioavailability of an active substance on safety and efficacy. This may happen when a given active substance (‘drug A’) interacts with another active substance (‘drug B’) on the level of transporting enzymes. If, for instance, drug B has a P-glycoprotein (P-gp) inhibiting effect and drug A is excreted back into the intestinal lumen by P-gp after absorption, this interaction will lead to an increase in the blood concentration and the bioavailability of drug A. However, the efficacy of drug A hardly increases as a result of this interaction, since the efficacy was already at its maximum before the interaction with drug B occurred (already at the lower concentration the occupancy of receptors causing the therapeutic effect was complete). Conversely, more adverse effects of drug A will occur, because of the increased blood levels leading to an increased occupancy of those receptors that are causing the adverse effects
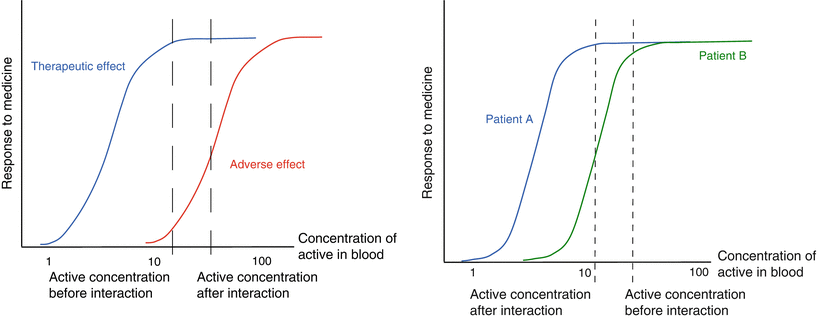
.
Fig. 16.3
Relationship between response to a medicine and blood concentration. The left figure shows the effect of an increased absorption on efficacy and safety. The right figure shows the effect that variation in bioavailability may have in patients with different sensitivity for an active substance
In Fig. 16.3 (right panel) the effect of a change in bioavailability is shown in relation to individual differences in drug sensitivity. The drug blood concentration versus effect relationships of the same active substance in two different patients are given in this figure. Patient A is more sensitive to the active substance than patient B. Due to an interaction between the medicine and food less active is absorbed (e.g. the effect of milk on tetracycline) in both patients. The bioavailability is reduced and the lower blood concentrations are reduced by about 30 %. For patient A this will not result in a significant change in efficacy. For the less sensitive patient B however, this interaction will lead to a more than 50 % reduction in efficacy. Such a reduction may, for example, lead to the emergence of drug resistant bacteria in the case of antibiotic therapy.
16.1.4 Solubility, Dissolution and Partition Coefficient
For many medicinal products dissolution in an aqueous environment (e.g. the fluids of the gastro-intestinal tract or mucosal lining fluids in the airways) is the major release mechanism of the active substance. The dissolved concentration of the active substance is the driving force for all diffusion-based drug transport mechanisms, since only the dissolved active substance is able to pass the absorbing membranes. Therefore the aqueous solubility of an active substance and the dissolution rate of the active substance from a medicinal product are considered to be highly relevant characteristics. To a large extent they determine the performance of a medicinal product in terms of bioavailability, efficacy and safety. The fundamentals of solubility and dissolution (rate) are described in Sect. 18.1.
The solubility of an active substance and the dissolution rate of a medicinal product can be varied by changing either the characteristics that relate to the active substance (such as particle size, salt form, crystalline form, the use of a pro-drug like an ester) or characteristics of the medicinal product (such as the use of disintegrants, complex-forming agents like cyclodextrins or polymers that form highly viscous gels, and the application of diffusion limiting coatings). See the box for examples.
There are numerous examples of medicinal products in which dissolution rate enhancing technologies are applied to increase the bioavailability or absorption rate of an active substance. Cardiac glycosides should be given as micronised particles in a solid oral dosage form because otherwise their dissolution rate and hence their bioavailability is too low. Piroxicam was shown to be absorbed faster when given as a cyclodextrin complex, which increases the dissolution rate. Similarly, the bioavailability of albendazole as a cyclodextrin complex was increased compared to crystalline non-complexed albendazole, based on the same mechanism. And finally, the bioavailability of amorphous chloramphenicol is higher than that of crystalline chloramphenicol.
Biological factors may change the solubility or dissolution rate of the active substance from the medicinal product as well. The residence time in the stomach may increase the dissolution of poorly water-soluble active substances and change their bioavailability. The pH in the stomach or the intestine may influence dissolution rates of acidic or basic active substances whose solubility is pH-dependent. Bile salts may increase the dissolution rate and thereby the absorption of poorly water-soluble active substances such as ciclosporine, phenytoin, levothyroxine and tacrolimus. Though, it has been shown that the association with bile acids reduces the absorption of the hydrophilic beta-blocker atenolol.
It is not only the solubility in aqueous solutions that may affect the biopharmaceutical behaviour of an active substance. The solubility in non-polar solvents is of importance too. The solubility in a lipid phase is of relevance to the passive transport of the substance over lipid membranes, a process that plays an important role in the absorption of many drugs. In order to quantify the lipophilicity of an active substance in relation to its aqueous solubility the concept of the partition coefficient was developed. The partition coefficient is defined as the quantitative distribution ratio of a dissolved substance over two immiscible liquids at equilibrium. In the pharmaceutical sciences the ratio of the concentrations in an aqueous phase (water or aqueous buffer solutions) and a lipid phase (e.g. n-octanol) is often considered. For this purpose the so called log P value has been defined. The partition coefficient between an aqueous and a lipid phase (log Po/w value) of non-ionised substances is defined according to (16.1):
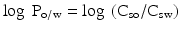
In this equation Csw is the saturation concentration of the active substance in the aqueous phase and Cso is the saturation concentration in the lipid phase. In general, the partition coefficient between an aqueous buffer or water and n-octanol is determined.
(16.1)
For active substances that can be ionised the distribution coefficient (log D) has been defined. The oil-water distribution coefficient (log Do/w value) for a substance is presented in (16.2):
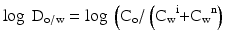
In this equation Co is the concentration of the substance in the lipid phase, Cw i is the concentration of the ionised substance in the aqueous buffer at a specific pH and Cw n is the concentration of the non-ionised substance at the same pH. From the definition it follows that the distribution coefficient varies with the pH of the aqueous buffer.
(16.2)
16.1.5 Absorption and Bioavailability
Poor aqueous solubility or a low dissolution rate or both may be the cause of poor bioavailability. For poorly water-soluble active substances, the amount and composition of endogenous aqueous fluid, present at the site of absorption will play an important role. In the stomach and in the small intestine there is ample aqueous fluid available, but in the mouth, the nose or the rectum a volume of only a few millilitres is available.
16.1.5.1 Absorption
After being dissolved the next step is the absorption, so passing the biological membrane. The surface area and type of membrane are major determinants for the extent and rate of absorption. Active substances may be transported over the membrane passively or actively. Absorption via passive transport may occur as paracellular transport through the interstitial spaces between the cells lining the absorptive membrane. For absorption by passive diffusion across a biological membrane, the concentration gradient is the driving force according to Fick’s diffusion law. A high external (or luminal) concentration (dissolved active substance at the site of absorption), will increase the absorption rate over the biological membrane. The continuous blood flow in combination with protein binding of the active substance will keep the internal concentration low and maintain the concentration gradient.
This transport mechanism is typical for the absorption via mucosal membranes such as the intestinal tract, the intra-oral (sublingual or buccal), nasal or pulmonary membranes. The size of the interstitial spaces in mucosal membranes may vary in size between around 0.4 nm for the duodenum up to about 4 nm for the alveolar membranes in the lung. In general the absorption through the interstitial space is limited to small hydrophilic molecules such as nitroglycerin after sublingual administration. The absorption of smaller proteins (up to a molecular weight of 20 kDa) via the alveolar membrane in the lung is an exception to this rule. The larger interstitial spaces between the alveolar type I cells (covering 93 % of the alveolar surface) enable what may be the only non-parenteral route for the administration of proteins.
Passive transport of lipophilic substances may occur via the fluid bilayer membrane of the cells lining the membranes. This transcellular route is the most important route for membrane passage of active substances. This transport mechanism is not only driven by the concentration gradient over the absorptive membrane, but also by the oil to water partition coefficient of the active substance (expressed as the octanol-water partition coefficient, which describes the ratio of the substance’s solubility in aqueous and fatty phases, see Sect. 16.1.9). More lipophilic substances will be transported faster over the lipophilic absorption membrane than those with a more hydrophilic character. In general, lipophilic active substances will not accumulate into the lipid parts of the absorption membrane because the continuous blood flow in combination with protein binding of the active substance will keep the internal concentration low. They will also maintain the concentration gradient at a maximum causing rapid uptake of the substance into the systemic circulation.
The lipophilicity of an active substance, and thereby its tendency to pass lipophilic membranes, can be described in terms of the octanol-water partition coefficient and the polar surface area. The octanol-water partition coefficient, the log P value, should be within the 0.4–5.6 range and preferably below 3. The polar surface area of a compound is the total sum of the surface of the polar atoms in the molecule. In general this will refer to the oxygen and nitrogen atoms in the molecules and include the hydrogen atoms attached. Co-inclusion of sulfur and phosphor atoms in the calculations was shown to add little to the predictive value of the results. When the polar surface area of a molecule exceeds 140 Å2 the molecule is too hydrophilic and considered to be unsuitable for absorption after oral administration. For special barriers such as the highly lipophilic blood brain barrier the threshold is 60 Å2.
Apart from the partition coefficient and the polar surface area, several other molecular characteristics predict membrane permeation; major determinants are:
Molecular weight, which is preferably below 300–500 Da
Number of H-bond donors and H-bond acceptors, not more than 3 donors and not more than 3 acceptors
Number of rotatable bond bonds, not more than 3
Number of non-hydrogen atoms
Furthermore, a limited number of active substances are absorbed by active transcellular transport mechanisms, a process that may even occur opposing a concentration gradient. This mechanism is characterised by the involvement of transporter enzymes able to transport the molecules of the active substance over the intestinal membrane. An example is the absorption of angiotensin converting enzyme inhibitors such as lisinopril and enalapril, which is mediated by the di/tri-peptide transporter protein (PepT1). Competition for or induction of transport enzymes may have a significant effect on the absorption of these active substances. This makes them sensitive for drug-drug or drug-food interactions that may significantly affect the drug absorption, and thereby drug efficacy and safety.
16.1.5.2 Bioavailability
The bioavailability of an active substance is defined as the fraction of the administered active substance that enters the systemic circulation (for oral administration the circulation beyond the liver) in unchanged form. The absolute bioavailability of an active substance is calculated from the area under the curve (AUC) of the blood concentration of unchanged drug versus time profile found following intravenous administration (by definition 100 % bioavailability) and the AUC found following the chosen administration route, corrected for the dose (D). The bioavailability after, for example, oral administration (For) is thus calculated from (16.3):
![$$ {\mathrm{F}}_{\mathrm{or}} = \left[\left({\mathrm{AUC}}_{\mathrm{or}}.\ {\mathrm{D}}_{\mathrm{iv}}\right)/\left({\mathrm{AUC}}_{\mathrm{iv}}.\ {\mathrm{D}}_{\mathrm{or}}\right)\right]\ .\ 100\% $$](/wp-content/uploads/2017/01/A315000_1_En_16_Chapter_Equ3.gif)
Differences in bioavailability of different medicinal products or after administration via different routes can also be deduced from the ratio of the AUCs of the blood concentration versus time curves.
(16.3)
As well as the total absorbed fraction, the absorption rate is also an important aspect of bioavailability. The absorption rate is characterised from the maximum blood concentration and the time at which the maximum concentration occurs in the concentration versus time curve.
The absorption rate of an active substance is relevant to the therapeutic effect for various reasons. If an active substance is used in an acute situation (e.g. epileptic insult, asthmatic attack, sleep induction, pain killing), a high absorption rate is preferred. A formulation design in which the active substance is already dissolved, may be useful in such case. A prolonged effect can be achieved however by a dosage form from which an active substance is released slowly (sustained release). Finally, if an active substance has a longer residence time in the absorbing organs, the bioavailability may be negatively influenced by, for example, enzymatic or chemical degradation.
16.1.5.3 Dose Number and Biopharmaceutical Classification System
The solubility of an active substance in the different body fluids and the efficacy of the absorption process, together with the dose determine the bioavailability of a medicine. The dose number (DN) is a dimensionless parameter that links solubility (Cs) to dose (D) and volume available for dissolution (V) during the absorption process. The dose number can be calculated from (16.4):
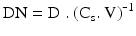
When the dose number is less than 0.1 dissolution is not expected to affect the absorption process, whereas at a dose number above 10 dissolution is most likely to decrease the absorption (rate) of an active substance. In general, solubilisation technology has to be applied for such compounds. When the dose number is between 0.1 and 10 the effect of dissolution on drug absorption should be investigated, to find out whether the bioavailability is affected by the dissolution behaviour.
(16.4)
The corticosteroid dexamethasone has a poor aqueous solubility of 89 microgram/mL. It is given by the oral route for different diseases. When the medicine is given with a glass of water the oral route offers a dissolution volume of about 500 mL in the stomach. When the 4 mg dose used in rheumatic diseases is given by the oral route the dose number will be 0.09 and no major dissolution problems are to be expected. However, when dexamethasone is used in pyodermia gangrenosum the dose has to be 300 mg. At this dose, the dose number will be 6.7. This is close to 10 and the dissolution behaviour of the medicinal product has to be investigated in order to prevent incomplete absorption of the dexamethasone.
The biopharmaceutical classification system (BCS) not only considers solubility but also considers the permeability of the absorbing membrane. This system classifies active substances based on their water-solubility in relation to their dose and the route of administration and to their permeability over the absorptive membranes. The BCS divides active substances into four classes:
Class I: high solubility, high permeability
Class II: low solubility, high permeability
Class III: high solubility, low permeability
Class IV: low solubility, low permeability
This classification system is used to characterise potential problems related to bioavailability and bioequivalence of an active substance [1].
The variable solubility is determined by the intrinsic solubility of the active substance in the aqueous fluids available for dissolution, the dose at which the medicine is given and the volume of liquid that is available for a certain route of administration. A medicine that is administered orally will have a larger volume of liquid available (250–750 mL) for dissolution than a medicine that is administered via the sublingual or rectal route (5–15 mL) and the composition of the different fluids varies. As a result, dissolution problems are to be expected for the sublingual or rectal route at lower doses than for the oral route. In general an active substance is considered to be highly soluble when the highest dose applied dissolves in about one third of the total available volume for dissolution. For the oral route, for example, an active substance is considered highly soluble when the highest dose dissolves in 250 mL of an aqueous liquid with a pH between 1.0 and 7.5.
The variable permeability is determined by the absorption of the dissolved active substance over the membrane. This may be measured by the direct assessment of the mass transfer (rate) over the human intestinal membrane or predicted from relevant animal models or in vitro epithelial cell culture models. An active substance is considered highly permeable when the extent of absorption is 90 % or higher.
Knowledge of the bioavailability and absorption rate of an active substance is important because these are important determinants of efficacy and safety. Large variations in bioavailability or absorption rate may, in the case of decreased absorption (rate), result in insufficient efficacy. In the case of unexpectedly high absorption it may lead to toxicity or serious adverse effects. Quantitative data on the bioavailability and absorption rate are necessary to evaluate the equivalence or non-equivalence between different medicinal products with the same active substance. These data may be used as surrogate parameters to establish efficacy and safety of a (generic) product.
According to the EMA two products are considered to be bioequivalent when they contain the same active substance and when their respective bioavailabilities (rate and extent) after administration in the same molar dose and via the same route, lie within acceptable predefined limits. These limits are set to ensure comparable in vivo performance, i.e. similarity in terms of safety and efficacy. The design and number of studies that is to be carried out to establish bioequivalence depends on the physico-chemical and pharmacokinetic properties of the active substance. In this respect reference is made to the BCS classification of the active substance. For BCS class I active substances it may even be possible to obtain a waiver for the in vivo studies (a so-called biowaiver), whereas for the active substances showing more complex pharmacokinetic behaviour extensive studies are to be carried out. In general bioequivalence will be determined from the parameters Cmax and AUC. Two products are considered to be bioequivalent when the 90 % confidence interval of the ratio of test and reference product falls within the 85–125 % acceptance interval. However, for the required design of the bioequivalence study and statistical evaluation details for a specific active substance reference is made to the appropriate (most recent) guideline on this subject [2, 3].
When in daily practice substitution (e.g. by a generic product) is considered, bioequivalence is of course the first parameter to evaluate. However, other aspects of medicine’s use related to both the product and the condition of the patient are to be taken into consideration as well. Among these are:
For active substances that have a narrow therapeutic window or non-linear kinetics substitution is not advised since even the smallest variations in the product and its performance may result in significant fluctuations in effect or safety. Examples of such active substances include: ciclosporin, tacrolimus, digoxin, ergotamine, levothyroxine, capecitabine and phenytoin.
Safety issues to be considered are the exact equivalence of the products (especially for ‘biologicals’ this may pose a problem) and the risk of allergy or intolerance for a specific excipient (e.g. colouring agents).
When specific administration devices (affecting parameters of relevance to the performance characteristics such as efficacy and safety of a medicinal product) are used substitution should not occur. Different dry powder inhalers, for example, generate aerosols of significantly different particle size. As a result, relevant variations may occur in lung deposition of the active substance. Therefore substitution of these types of products (e.g. dry powder inhalers) should not occur.
The appearance of the product. When large differences in appearance between both products exist, substitution may confuse the patient.
The performance characteristics and handling of both medicinal products should be similar (e.g. for auto-injectors, or the occurrence of a score on the tablet that allows for dividing a dose).
Medicinal products administered at a specific site to obtain a local effect should preferably not be absorbed systemically. However, significant amounts of active substance can be absorbed, e.g. after application on the skin. Removal of locally acting active substances from the site of action by systemic absorption may result in systemic effects that can be considered as adverse effects. After nasal, ocular, pulmonary and rectal administration of active substances for a local effect, absorption into the systemic circulation is likely to occur. This may cause adverse effects and limit the duration of the desired drug effect. Conversely it should be realised that the systemic route is often also the main route for clearance of the active substance from the site of administration. The bioavailability of locally acting medicines is, of course, not determined by the amount of active substance that reaches the systemic circulation. As an alternative the fraction of the active substance that is dissolved in the aqueous fluids at the site of application is usually taken as a measure for the bioavailability.
16.1.6 Excipient and Food Interactions
An active substance, although initially released from its dosage form (and dissolved), may become unavailable for absorption due to reactions with other medicines or food components [4]. An example is the formation of insoluble complexes of tetracycline with calcium or aluminium ions from antacids or milk products. Interaction (chelation or binding) with iron ions leads to a reduced absorption for a variety of active substances such as doxycycline, penicillamine, methyldopa and ciprofloxacin. The absorption of active substances showing pH-dependent dissolution behaviour may be influenced by medicines that influence the gastric pH, such as H2-antagonists, proton pump inhibitors and antacids. Antimycotic active substances such as ketoconazole or itraconazole dissolve better in acidic fluids. Therefore their bioavailability may be increased by the concomitant use of an acidic drink like cola, whereas the concomitant use of antacids or proton pump inhibitors is likely to reduce the bioavailability. Concomitant use of milk may increase the dissolution of acidic active substances, whereas fats from food may increase the bioavailability of lipophilic active substances like albendazole and griseofulvin.
In dermal preparations ion-pair formation between ionic surfactants and an active substance may reduce the local availability of the active substance. An example is the interaction between the anion laurylsulfate (from the surfactant sodium laurylsulfate) and the cationic neomycine or tetracycline in creams.
It is important that the (clinical) pharmacist always provides appropriate advice to the patient about how to take a medicine and warns against the concomitant use of other medicines or certain foods, in cases where interactions may occur. The potential risk of interactions may even be of greater clinical importance when they occur at the level of the metabolism of the active substance. This issue is discussed in Sect. 16.1.13.
16.1.7 Stability of the Active Substance in the Physiological Environment
Several active substances are unstable in an acidic environment and will degrade when in contact with gastric juice. Examples are omeprazole, pantoprazole, erythromycin and pancreatic enzymes. Such active substances are formulated in a tablet or pellets covered with an acid-resistant layer, a so-called enteric coating. The acidic nature of the polymers used in these coatings prevents dissolution in the gastric environment. The coating will dissolve in the small intestine (with higher pH values), after which the active substance is released. Alternatively, the active substance may be used in the form of a more stable salt, for example erythromycin ethylsuccinate.
Enteric coated products exist in various presentation forms. They include tablets surrounded with the coating, coated pellets in a capsule or coated pellets compressed into a tablet (see Sect. 4.10). This last form sets specific requirements to the quality of the coating around the pellets since the integrity of this coating must not be compromised during the tablet compaction process. Depending on the presentation form, tablet breaking may or may not be allowed. Tablets covered with an enteric coating may never be broken, whereas controlled release tablets produced from coated pellets may be divided or sometimes even dispersed for a short period of time in liquids or semisolid food (custard, yoghurt). The same can be done with capsules containing coated pellets. However, coated products should never be longer than 5 min in drinks or food before use. Crushing is not allowed for any controlled release formulation.
Metabolic instability of the active substance in the physiological environment may reduce bioavailability especially after oral administration. Enzymatic degradation of an active substance in the lumen of the stomach or intestine may reduce the amount of active substance available for absorption, whereas after absorption enzymes in the intestinal wall and the liver may reduce the bioavailability. This is called the first-pass effect.
After intramuscular or subcutaneous injection, bioavailability may become reduced because of enzymatic breakdown of the active substance at the site of injection.
16.1.8 First-Pass Effect
Following oral administration and absorption from the intestine, an active substance passes the intestinal wall and the liver. During uptake from the gastro-intestinal tract, enzymes present in the intestinal lumen or in cells of the intestinal wall can metabolise an active substance. Subsequently the absorbed active substance is transported to the liver by the portal vein system. During the first passage through the liver, liver enzymes may metabolise another part of the active substance. The metabolic degradation during the first passage of intestine and liver may significantly reduce the bioavailability since it occurs before the active substance is distributed over the entire body. This particular loss of active substance, after absorption but before distribution, is called the first-pass effect.
The extent of a first-pass effect of an active substance depends on the dose and absorption rate in relation to the enzymatic capacity. Moreover, the first-pass effect can be influenced by the concomitant use of food, which may affect the absorption rate or induce or inhibit the enzymatic degradation of certain active substances. From this perspective it is clear that the bioavailability of an active substance can be optimised by taking a medicine at the appropriate time, e.g. before, during or after a meal. Examples of active substances that undergo a high first-pass effect after oral administration include: amiodarone, ciclosporin, 6-mercaptopurin, metoprolol, midazolam, morphine, nifedipine, propranolol, saquinavir, tacrolimus, terbutaline and verapamil.
The hepatic first-pass effect can be circumvented by giving a medicine through a route of administration that does not primarily absorb the active substance via the portal vein system, such as an intramuscular or subcutaneous injection, sublingual or nasal administration or via the pulmonary route. The rectal route is often mentioned as an option too. However, since two of the three rectal veins do end up in the portal vein, the maximum effect of this route will be a reduction of the first-pass affect by only about 30 %.
16.1.8.1 First-pass metabolism and controlled release products
In order to understand the relevance of first-pass metabolism for the formulation of controlled release products, one should have a basic understanding of enzyme kinetics. Enzyme kinetics are described using the Michaelis-Menten equation (16.5). This equation describes the rate of transformation of a substrate (the active substance) by an enzyme.
![$$ \upnu = {\mathrm{V}}_{\max }.\left[\mathrm{S}\right]/\ \left({\mathrm{K}}_{\mathrm{m}} + \left[\mathrm{S}\right]\right) $$](/wp-content/uploads/2017/01/A315000_1_En_16_Chapter_Equ5.gif)
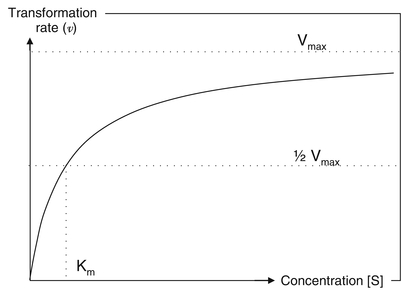
In this equation ν is the enzymatic reaction rate, Vmax represents the maximum transformation rate (the rate achieved at complete saturation of the enzymes), [S] is the substrate concentration and Km the substrate concentration at ½Vmax. Figure 16.4 shows the relationship between the substrate (active substance) concentration and the transformation rate.
(16.5)
Fig. 16.4
Relationship between the substrate concentration [S] and rate of transformation [ν]
At low concentrations an enzymatic system is able to efficiently break down the presented active substance. When the concentration of active substance (substrate) rises, the transformation rate will increase proportionally to the increase in concentration of active substance. As a result the relative amount of active that is metabolised will remain the same. However, as the concentration increases further (significantly beyond the Km) the enzymatic system will get saturated and the increase in rate of transformation will no longer be proportional to the increase in concentration. As a result relatively more active substance will escape from transformation by the metabolic enzymes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


