Biology of Cervical Squamous Neoplasia
Introduction
In the past half-century, cytologic screening, particularly in high-resource settings, has dramatically decreased the burden of squamous cervical cancers, virtually all of which are caused by tissue-specific, persistent infection with human papillomavirus (HPV). The elucidation of the infectious etiology of this disease led to a Nobel Prize in 2008, and to development of preventative vaccines that became commercially available in 2006. This was a public health milestone; worldwide, chronic infections initiate approximately 20% of human cancers,1 with HPVs causing more malignancies than any other virus. However, because primary prevention strategies are cumbersome and expensive, cervical cancer is still the third most common cause of cancer death in women.2 Moreover, the incidence of HPV-associated cancers in other anatomic sites, for which no screening algorithms have been developed, is increasing rapidly.
HPV disease is not restricted to the cervix. A growing body of evidence links persistent HPV infection to squamous cancers of the vagina, vulva, anus, and oropharynx.3 Almost all vaginal and anal squamous cancers4 and approximately 20% of squamous cancers of the vulva5 are caused by HPV. The incidence of preinvasive vulvar disease attributed to HPV has increased by 411% over the past three decades, primarily in women under the age 65.6 HPVs have been identified in one in four squamous carcinomas of the head and neck; to date, the incidence of HPV-associated oropharyngeal cancers is greater in men than in women.7 Indeed, in the United States, the incidence of HPV-associated cancers of the oropharynx is approximately equal to that of cervical cancer, and is likely to surpass it soon.8 Because prophylactic vaccines are not administered to the full target population, cohort-appropriate strategies for preventing and treating HPV disease are needed. A better understanding of the biology of intraepithelial, preinvasive HPV disease will inform strategies for screening, secondary prevention, and treatment.
HPV Infection
Like other viruses that cause human cancers, most HPV infections are asymptomatic, eventually cleared, and do not harm the host (Figure 9.1). HPV is an epitheliotropic virus that is essentially endemic. In unvaccinated cohorts, the lifetime risk of acquiring genital HPV at least once is greater than 80%.9 Over 40 genotypes of HPV can infect the genital tract, 15 of which have been classified as carcinogenic (Table 9.1). These include HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, and 72.10 Together, HPV 16 and HPV 18 are the most common carcinogenic genotypes worldwide (Figures 9.2 and 9.3).11
Table 9.1
Classification of HPV Types Associated with Genital Lesions
| Classification | HPV Type |
| Oncogenic | 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, 82 |
| Putatively oncogenic | 26, 53, 66, 70 |
| Non-oncogenic | 6, 11, 40, 42, 54, 55, 57, 84 |
Adapted from Muñoz et al.13
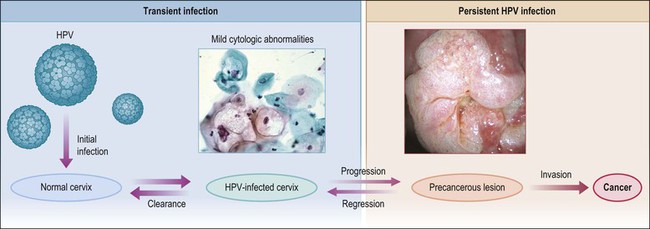
Figure 9.1 Model of HPV-induced cervical pathogenesis, proceeding from HPV infection of the normal cervix to mild cytologic abnormalities, progression to persistent HPV infection and precancerous lesions, and, finally, invasion. Of significance is the ability for precancerous lesions to regress, and the ability of most HPV-infected cervices to clear infection. (Reproduced with permission from Wright and Schiffman.118)
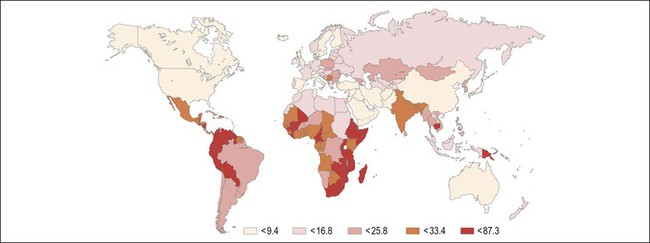
Figure 9.2 Age-standardized incidence rate of cervix uteri per 100,000 women worldwide in the year 2002. The lowest disease burden is largely found among regions with effective screening programs (e.g., North America) and regions where HPV prevalence is low (e.g., China). The greatest disease burden is denoted in red and is found in those countries with high HPV prevalence and without an effective screening program (e.g., East Africa). (Reproduced with permission from Ferlay et al.119)
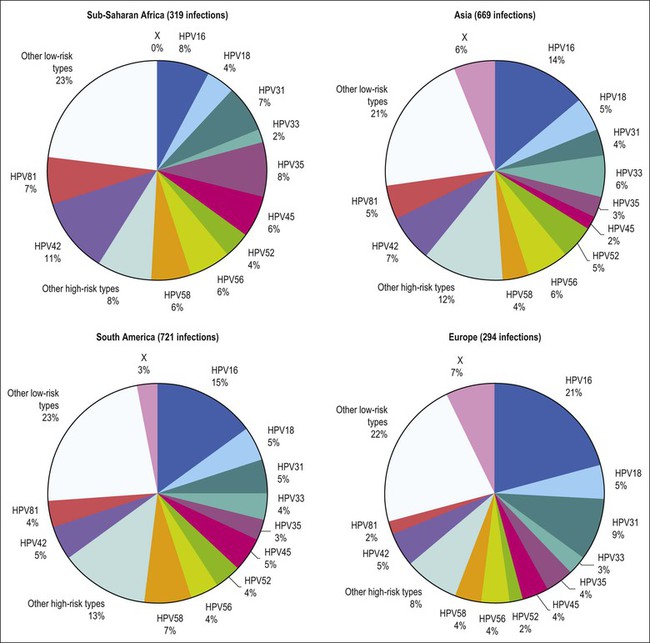
Figure 9.3 Prevalence of HPV types in cytologically normal women worldwide, by geographic region. HPV type-specific burden varies by geography, with HPV 16 prevalence highest (21%) in Europe but HPV 18 prevalence equivalent in all regions. (Reproduced from Clifford et al,120 with permission from Elsevier)
HPV infects cervical epithelium shortly after sexual debut, and can be detected in up to 50% of young women who have initiated sexual intercourse in the preceding 36 months.12 Most women clear an incident HPV infection within 1–2 years. Several host cofactors that have been associated with progression of HPV infection to carcinoma are shown in Table 9.2. Older women take longer to clear infections, as do smokers, and women with underlying immunosuppression.14 In the cervix, persistent infection is the proximate cause of squamous cancers and their precursors, high-grade squamous intraepithelial lesion (HSIL), or cervical intraepithelial neoplasia (CIN) grade 2 or 3 (CIN 2, CIN 3).
Table 9.2
Summary of Evaluated HPV Cofactors for Squamous Cell Carcinoma and Adenocarcinoma of the Cervix
| HPV Cofactor | Squamous Cell Carcinoma | Adenocarcinoma |
| Smoking | ++ Increased | No association |
| Parity | ++ Increased | No association |
| Oral contraceptives/hormones | ++ Increased | + Increased |
| Chlamydia | Increased, but current evidence not significant | No association |
| Herpes simplex virus 2 | Increased, but current evidence not significant | No association |
| Antioxidants | Decreased, but current evidence not significant | Current evidence not significant |
| Obesity | No association | + Increased |
HPV infections are limited to the suprabasal compartment of nonsterile barrier epithelia in which the immunologic contributions of the local microbiome are incompletely understood. Viral replication, assembly, and release of virions occur in the context of host cellular maturation and desquamation, without cell lysis, and without systemic viremia. Persistent HPV infections increase susceptibility to malignant transformation by a variety of mechanisms, including enhancing genetic instability and cell proliferation, inducing angiogenesis, interfering with intrinsic and extrinsic apoptotic pathways, downregulating expression of adhesion molecules, abrogating DNA damage responses, and interfering with both innate and adaptive immune responses in the lesion microenvironment. Known virally mediated immune-suppressive mechanisms include interfering with type I interferon responses and Toll-like receptor (TLR) signaling, downregulating cell surface MHC class I expression, enhancing secretion of immunosuppressive cytokines, recruitment of immune cells with suppressive function, and downregulating expression of adhesion molecules in lesional neovascular endothelium.15–17 Indeed, as we learn more about the immunobiology of persistent infection it becomes less surprising that immune-based therapeutic strategies to date, which have focused solely on eliciting an effector T-cell response to HPV antigens, have failed.
Low-Grade Squamous Intraepithelial Lesions (CIN1)
HPV infects keratinocytes at the basal epithelial layer of the squamocolumnar junction.18 A productive infection leads to the production of whole infectious virions, and is limited to epithelia that undergo maturation. As HPV does not encode DNA polymerase, host cellular DNA polymerase must be expressed for viral replication. In normal physiologic conditions in stratified squamous epithelium, DNA is copied only in the basal layer. In contrast, in virally infected epithelium, HPV drives DNA replication and cell division in the upper layers of differentiating epithelium, where HPV DNA is packaged into viral capsids, resulting in infectious virions (Figure 9.4).19 Infectious virions are released not by cell lysis, but in the course of host cell maturation and desquamation, without eliciting an inflammatory response.
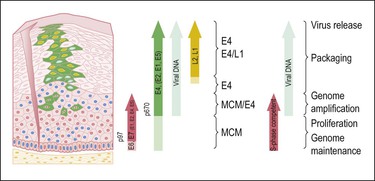
Figure 9.4 HPV gene expression during its life cycle in the genital squamous mucosa, from HPV-infected basal cell and maturation to surface of epithelium. Upon infection, the viral genome is maintained as a low copy number episome. During epithelial differentiation, the p97 promoter directs E6 and E7 expression for S-phase entry (red) and viral replication proteins (E1, E2, E4, E5) increase in abundance (green), facilitating amplification of viral genomes (blue). E4 persists in upper epithelial layers where viral capsid proteins (L1, L2; yellow) are found. (Reproduced with permission from Doorbar.121)
HPV infection produces a distinctive cytopathic effect that is recognizable both cytologically and histologically as a sharply demarcated cytoplasmic halo surrounding an enlarged, irregular, hyperchromatic nucleus with uneven chromatin distribution, termed ‘koilocytic atypia.’20 Other findings include multinucleation, parakeratosis, and hyperkeratosis. Histologically, HPV infections can be distinguished by koilocytotic changes in differentiating cells, with increased ratios of nuclear to cytoplasmic area, and mitotically active cells above the basal layer. Clinically, the behavior of cytology interpreted as ‘atypical squamous cells of undetermined significance’ with a concurrent positive test for oncogenic HPV DNA, cytology interpreted as ‘low-grade squamous intraepithelial lesion,’ or a tissue biopsy diagnosis of ‘low-grade squamous intraepithelial lesion’ or ‘cervical intraepithelial neoplasia grade I’ (CIN 1) is essentially the same; most eventually regress without intervention. Most CIN 1 lesions maintain HPV as an episome, sustain a complete viral replication cycle, and can be thought of as HPV infection.
High-Grade Squamous Intraepithelial Lesions (CIN 2/3)
HSILs, or CIN grade 2 or 3 (CIN 2/3), are associated with integration of the viral genome into the host genome, and subsequent constitutive expression of the E6 and E7 viral proteins (Figure 9.5). These proteins are functionally required for initiation and persistence of disease.21,22 Viral integration sites, although randomly distributed within the human genome,23 occur principally at sites where human DNA is prone to breakage (e.g., fragile sites), and appear to affect only the expression of the HPV genome itself. Specifically, during integration, E1 and/or E2 are frequently disrupted, while the E6 and E7 viral oncogenes are retained, resulting in constitutive expression. Morphologically at the cellular level, HSILs are characterized by a high nuclear to cytoplasmic ratio. Histologically, high-grade lesions display disarray of the basal layer itself, full thickness lack of cell maturation, and are mitotically active.
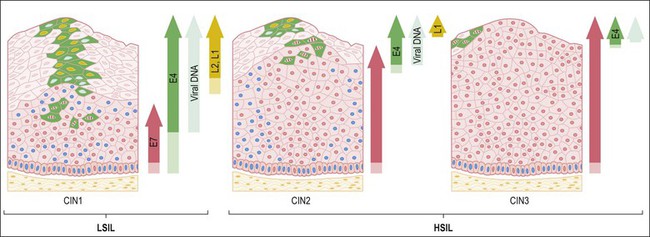
Figure 9.5 HPV early and late gene expression from initial HPV infection through maturation, from CIN 1–3, by evolutionary HPV type. The productive cycle begins close to the basal layer and viral genome amplification begins in the parabasal cell layers. (Reproduced with permission from Doorbar.121)
Although all squamous cervical cancers arise from untreated HSIL, not all HSILs progress to invasive cancer. Approximately 35% of HSILs undergo complete regression in a time frame of 4–6 months.24,25 Lesions associated with HPV 16 are less likely to regress than lesions associated with other HPV types. Because it is not possible to distinguish HSILs that are likely to regress from those that are not, the standard of care is excision or ablation. Excisional approaches include loop electrocautery excision procedure or cold knife conization. The rate of recurrence is 10%.26 Iterative excisional procedures are associated with cervical incompetence and subsequent preterm birth.27,28
Mechanisms of Oncogenesis
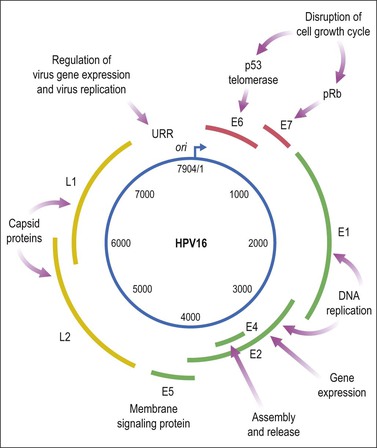
HPV is a small, double-stranded DNA virus with an 8 kb genome comprised of three domains (Figure 9.6): a noncoding upstream regulatory region, an early region containing six genes (E1–E7), and a late region encoding capsid proteins L1 (major capsid) and L2 (minor capsid).29 The E6 and E7 viral proteins enhance cell proliferation in the face of abrogated DNA damage responses and genetic instability. Additionally, E6 mediates immortalization by activating telomerase. Finally, these oncoproteins compromise both innate and adaptive immune responses in the tissue microenvironment.
Viral persistence is mediated in part by dysregulated innate immunity involving pattern recognition receptor, particularly alterations in expression and function of TLRs in dendritic cells and macrophages. However, as TLRs sense pathogen-associated molecular patterns, including viral genetic material, they are also found in normal cervical epithelial cells.30,31 While less is known about TLR function in epithelial cells, evidence of dysregulated TLR3 signaling, including dampened expression of genes encoding antimicrobial molecules, chemotactic and pro-inflammatory cytokines, and proteins involved in antigen presentation, has been identified in cervical keratinocytes containing episomal HPV.32
The HPV E6 and E7 Oncoproteins
E6 binds to an endogenous E3 ubiquitin ligase, E6 associated protein (E6AP).33 The E6/E6AP heterodimer binds p53, a tumor suppressor protein that also regulates apoptosis, and targets it for rapid proteasome-mediated degradation.34 p53 can be stimulated either by genotoxic or cytotoxic damage, or by dysregulated DNA synthesis. These signals would be triggered in an HPV infection, particularly in a persistent infection in which cell cycling continues despite cellular damage.
In infected cells, E6 blocks both the intrinsic and extrinsic apoptosis pathways.35 The extrinsic apoptosis pathway is triggered by viral infections and activates death receptors on the cell surface. E6 blocks death receptors from interacting with death-inducing signaling complexes, and also degrades signaling proteins, such as FADD and caspase-8.36,37 The intrinsic apoptosis pathway is activated by DNA damage and oxidative stress. The intrinsic pathway starts in the mitochondria with Bak, and Bak expression in the mitochondrial membrane is blocked by E6.38 Downstream proteins, such as additional caspases, are degraded by E6, and inhibitors of apoptosis, including NF kappa B, survivin, and c-IAP, are stabilized by E6.39,40
E6 also has direct roles in chromosomal instability and DNA damage. Minichromosome maintenance 7 (MCM7) insures there is only a single round of DNA replication in a cell cycle. E6 degrades MCM7, allowing multiple copies of its own DNA to be made.41 A byproduct of HPV DNA amplification is the risk of excessive cellular chromatin duplication. This aneuploidy normally would be sensed in G2 by p53, triggering cell cycle arrest and apoptosis, but p53 is removed by E6/E6AP. E6 also degrades XRCC1 and O6MGMT.42,43 Both are involved in single-stranded DNA repair, and, without them, stochastic errors that drive cancer development can accrue.
E6 disrupts replicative senescence signals in keratinocytes. With each copy, linear chromosomes lose 100–200 bases of repetitive telomeric DNA. Thus, the age of a cell is inversely proportional to the length of its telomeres. Replicative senescence occurs when somatic cells recognize their intrinsic age.44 Stem cells express telomerase, a ribonucleoprotein, to extend this DNA, and the catalytic subunit of telomerase, hTERT, is rate determining.45 By lengthening telomeric DNA, stem cells avoid cellular senescence. Almost all cancers express telomerase to also avoid replicative senescence signals.46 E6/E6AP enables replicative immortality through hTERT activation. E6/E6AP removes repressors and recruits activators to the normally constitutively repressed promoter of hTERT, and E6 also affects the stability of hTERT mRNA to increase protein production and telomerase activity.47–52
E6 degrades proteins involved in cell–cell adhesion and apicobasal polarity. It degrades hScrib, a tight-junction protein, and many postsynaptic density protein 95, drosophila disc large tumor suppressor, zonula occludens-1 protein (PDZ) domain-containing proteins.53–57 Finally E6 binds proteins in the extracellular matrix.58 These changes are important to allow viral release from cells as they slough off the upper layers of stratified squamous epithelium. They are also important in invasion and metastases.59,60
Finally, E6 interferes with innate immune responses. Interferon regulatory factor 3 (IRF-3) is normally activated by a viral infection, but is inhibited by E6.61 TLR9 is activated by viral DNA motifs and induces cytokine production, but E6 blocks its expression.62
The second crucial HPV oncoprotein, E7, destabilizes retinoblastoma protein (pRb) and other pocket proteins by targeting them for proteasome-mediated degradation, resulting in cell cycle progression, as these proteins normally hold cells in G1, not allowing DNA synthesis.63–65 In E7-driven proliferation, S-phase gene expression ensues, and viral DNA is copied in addition to cellular DNA. E7 drives cell division gene activation and increases the open chromatin structure at gene promoters. E7 also binds histone deacetylases and represses polycomb group complexes, some of which are histone methyltransferases.66,67
E7 not only pushes cells to continue to divide, but also helps them to evade growth suppressors. E7 blocks transforming growth factor-beta (TGF-β) and tumor necrosis factor (TNF)-alpha G1.68,69 TNF-α G1 is a cytokine that induces programmed cell death, and is produced by cytotoxic T-cells to eliminate virally infected cells. Activation of IRFs is also blocked by E7, thereby compromising activation of innate immunity.70
E7 also binds to p600, a pRb-associated factor critical for anchorage-dependent growth.71 E7 increases expression of vascular endothelial growth factor and IL-8, both of which are angiogenic, and enhances transcriptional activity of hypoxia inducible factor 1α (HIF 1α), and disrupts interaction with histone deacetylases.72–74 Finally E7, like E6, can induce DNA damage at the chromosomal level. E7 increases centrosome amplification early in the viral life cycle.75,76 This increase in centrosomes can lead to DNA damage, and increases the frequency of DNA integration. As has been demonstrated in cell lines and mouse model systems, E6 and E7 (or equivalently inactivation of p53 and Rb) cooperate to produce full-fledged malignancy.77
Naturally Occurring Immune Responses to HPV Antigens
In immune-competent hosts, over 90% of genital HPV infections become undetectable without intervention.78 However, naturally occurring systemic humoral and adaptive responses to HPV antigens, even in cohorts with documented type-specific mucosal infections that have become undetectable, are hard to detect in peripheral blood.79–81 Type-specific serum antibodies to capsid proteins are detectable in less than half of women in whom cervical HPV infections of known serotype have cleared. Nonetheless, data from cohorts undergoing prophylactic vaccination demonstrate an anamnestic response to a single dose of virus-like particle vaccine in previously infected subjects.82 Dysplasia is associated with ineffective immune responses to viral nonstructural proteins. Antibody to E7 can be measured in persons with invasive cancer, but not in earlier stage disease.83
HSIL (CIN 2/3) is a lesion that should be susceptible to an HPV-specific effector immune response. Expression of viral, non-‘self’ proteins is functionally required to initiate and maintain the transformed phenotype, thereby providing true tumor-associated antigenic targets.21,22 Not all cervical HSILs progress to cancer; 25% of established CIN 2/3 lesions caused by HPV 16, the genotype most commonly associated with malignancy, undergo complete regression.25
However, women with squamous intraepithelial HPV lesions rarely have systemic T-cell responses to HPV E6 or E7 that can be detected directly ex vivo, likely reflecting the tissue compartmentalization of early disease. In contrast to immune responses to other viral infections, the frequency of systemic memory CD8+ T-cells in individuals with a known prior cervical HPV infection, which has subsequently become undetectable, is vanishingly low. For example, using direct ex vivo assays, the frequency of systemic virus-specific CD8+ T-cells after primary infection with cytomegalovirus or hepatitis C virus (HCV) can be up to 5%.84,85 In contrast, in subjects with CIN, the frequency of HPV-specific T-cells is two to three orders of magnitude lower, in the range of 0.1–0.01%.86 Detection of systemic HPV-specific T-cell responses in subjects with intraepithelial neoplasia requires ex vivo sensitization.87–89 After in vitro stimulation with HPV antigens, peptide-specific T-cell frequencies increase in subjects with concurrent disease at the time of blood sampling, compared with subjects with no evidence of disease.86,90 While amplification can identify qualitative responses to HPV antigens, it is likely to have limited use in accurately distinguishing quantitative differences between individual subjects, either in the course of a natural infection or in subjects with intraepithelial disease. Responses identified only after prolonged in vitro restimulation likely represent expansion of previously induced memory immune responses, rather than an ongoing response at the time of sampling.
In immune-competent persons with undetectable systemic HPV T-cell responses, dense immune cell infiltrates nonetheless localize at the lesion site, and, in persistent disease, fail to access the lesional epithelium. In contrast, dysplastic lesions that ‘permit’ CD8+ T-cell access to the epithelial compartment are likely to regress.17 This observation suggests that, even in the clinical setting of preinvasive, incipient neoplasia, factors in the lesions mitigate the ability of recruited CD8+ T-cells to eliminate disease.



