- Cancers may be cured and life expectancy improved by surgery and/or local radiotherapy, but only if the entire tumor is accessible and as long as viable cells have not already relocated to other sites.
- Once cancers have spread, excepting rare cases where single metastases can be successfully removed, hope of a cure rests on the success of systemic treatments. However, even where no cure is possible, localized treatments remain a viable means of alleviating symptoms in order to improve quality if not quantity of life.
- Drugs may be used alone or as “adjuvants,” given after surgery or before it (neoadjuvants) in order to improve long-term outcome by eradicating cancer cells that may already have escaped the primary site.
- With notable exceptions, such as testicular cancer and Hodgkin lymphoma, chemo- and radiotherapy treatments arrest but rarely eradicate the cancer, and the disease recurs or progresses.
- Treatment failure, through inherent or acquired resistance to drugs, is a major and common problem in cancer therapy. Thus, apparently successfully treated cancers can be repopulated by cancer cells that have endured the treatment either through their intrinsic resilience or by sheltering within oncogenic oases within the tumor, as exemplified by stem cell niches.
- Cancer stem cells (CSCs), a newly recognized subpopulation of “über”-cancer cells, may be very hard to kill from the outset, or new coteries of drug-resistant cancer cell clones may emerge during chemotherapy.
- More recently, cancer cells have been observed to enter a state of “dormancy,” in which they may survive treatments and from which they may unpredictably awaken at some future time. Reminiscent of trench warfare, a protracted and seemingly unsurvivable barrage of chemo-radiotherapy lays waste to the tumor. Yet, still the whistle eventually sounds and the battle-hardened survivors emerge from their dugouts and, from no-man’s land, sally forth and capture new positions.
- In fact, these post-apocalyptic cancer cells often prove remarkably resistant to further treatments. Thus, a pessimist might conclude that cytotoxic therapies directed against cancer cells, which only eliminate the camp followers, may simply clear the way for the expansion of more militant CSCs that have been left behind. The resultant revenant tumor, re-emerging phoenix-like from the ashes, will, compared to its forebear, be more aggressive, progress more rapidly, and be less sensitive to therapeutic attack.
- As was discussed in other chapters, the major limitation surrounding traditional chemotherapy is the extent of collateral damage suffered by normal cells, particularly those in bone marrow and epithelia. Thus the oncologist has to steer the patient between Scylla and Charybdis, between treatment failure and infection.
- It is often assumed that early diagnosis would improve the efficacy of most of our anticancer treatments. In particular, finding primary tumors that comprehend only stay-at-home cancer cells should enable treatment to be curative. Although likely to be true in most cases, this notion will be hard to prove until we achieve these aims and can actually test it.
- New analytical and imaging tools are under development that may allow us to pick up smaller and clinically silent tumors. However, we are a long way from being able to pick up the spoor of the first few cancer cells, at least until they have gone through a very large number of cell divisions. As each cancer cell division is a potential mutagenic event, such delays inevitably increase the chance that by diagnosis there will already be a vanguard of invading or metastasizing cancer cells, thereby precluding curative treatment.
- Progress in early detection and tracking down of cancer cells will offer little, if we lack the wherewithal to exploit this advantage.
- All too often, these ever resourceful foes manage to successively evade the finely honed edge of the surgeon’s knife, find shelter from the withering blasts of radiotherapy, and finally ford a raging torrent of chemotherapy to reach and colonize a distant shore. It is the researchers who must ensure that clinicians are continually re-armed and able to contend with or preempt the evolving survival instincts and adaptive capabilities of the cancer cell, and they have responded magnificently to the challenge.
- Thus, it may once have been true to say that for cancer patients nothing was certain except death and taxols, but times are changing fast. In many cases, cancer has been relegated from the premier league of rapidly life-threatening conditions to the lower divisions along with other chronic diseases. This raises a series of new challenges including how to support patients living long term with cancer, prolonged periods of monitoring, chemotherapy, and symptom control (Chapter 19).
- Subtle refinements notwithstanding, modern approaches to cancer drug development are turning away from traditional chemotherapies, which paralyze cell division or damage DNA, in order to concentrate almost exclusively on targeted agents that are aimed at cancer-relevant molecules as exemplified by the oncogenic tyrosine kinases, BCR–ABL, EGFR, and HER2.
- Together, the molecular aberrations in a tumor constitute the malignant manifesto of the cancer cell in which the varied aspirations for the future are clearly set out and the key processes required to achieve them are underscored. By aiming for these essential processes we might, by the judicious placement of a few well-designed therapeutic obstacles, thwart the pretentions of the cancer cell and the realization of its manifesto promise.
- The ideal treatment will have an absolute predilection for cancer cells and would be entirely innocuous to normal cells. Given that traditional chemotherapy is often limited by toxic effects on noncancer cells, the closer we get to this ideal of “sectarian” cytotoxicity, the greater the chance of successfully completing a course of treatment that will eradicate the cancer.
- Cancer cells often become overreliant on certain aberrant pathways to support growth or even survival, a situation referred to as oncogene addiction. Oncogenically jaded cancer cells have little room left to accommodate other growth-supporting signals, which are often irrevocably suppressed.
- This situation offers unique opportunities to effectively sabotage the entire malignant manifesto by deploying targeted agents directed only against these central supports of oncogenic growth. Such maneuvers are also much less likely to be generally harmful to normal cells, in which these addictive pathways are supernumerary or nonexistent, because the alternative compensatory options have not been suppressed.
- Pioneering fin-de-siècle successes in the 1990s, exemplified by the introduction of imatinib to target the abnormal BCR–ABL fusion protein in chronic myeloid leukemia, have validated the concept of biological targeting. The ensuing flurry of research activity has now delivered a profusion of similar and even wildly dissimilar agents into current clinical use.
- However, despite early promise, long-term benefits have been limited by the near-universal development of resistance of cancer cells to these molecular targeted agents.
- It is anticipated that understanding the mechanisms through which cancer cells circumvent the growth-inhibiting actions of targeted agents will in future allow combinations of agents to be deployed that will avoid or overcome resistance.
- In many cases, resistance arises when the therapeutic blockade is bypassed by further mutations, which activate the same or another growth-supporting pathway independently of the targeted protein.
- Some cancer-causing proteins, such as RAS, have proved enormously difficult to target with small-molecule drugs. A variety of strategies are being explored to contend with this challenge. These include targeting of essential, but more accessible, oncogenic partners, using new agents such as siRNA, or adopting entirely new conceptual approaches to therapy. One such is synthetic lethality, whereby addiction to a particular oncogene (or loss of a tumor suppressor) also renders the cancer cell addicted to some other protein or signaling pathway, exemplified by PARP in BRCA1-deficient breast cancer cells, which may prove easier to target.
- Recent studies are revealing a new and intriguing feature of cancer cells: for many oncogenic mutations, either too much or too little of a good thing may not do. Rather, just the right amount of a given oncogene may be needed in order to produce cancer cells with exactly the right blend of endurance, fecundity, and freedom of movement. Too much and the cancer may fail as powerful anticancer processes are brought to bear.
- Another promising approach is to employ treatments to specifically prevent the spread of cancer cells, which is after all the cause of most deaths from cancer. As long as cancer cells remain hobbled to the primary tumor, the prognosis will generally remain good, with the most obvious exceptions being brain tumors where the primary alone may shorten life. Once cancer cells begin their peregrinations and distant organs become home to thriving colonies of émigré cancer cells, however, the outlook rapidly deteriorates.
- Antimetastatic therapies remain in their infancy, a state of affairs that appears surprising given that the vast majority of cancers would prove nonlethal if their spread were curtailed. A significant barrier has been the difficulty in testing such agents in clinical trials – long follow-up and large numbers of patients would be required to prove an antimetastatic effect, and the cost would prove prohibitive when compared to using tumor shrinkage as an endpoint. Surrogate endpoints such as numbers of circulating cancer cells or markers of vascular invasion may be needed to address this problem in future.
- There are also concerns over the long-term security of antimetastatic or cytostatic therapies, which leave primary tumors in place. After all, how durable is cancer cell rehabilitation? Maybe for cancer cells, only complete obliteration can offer reassurance.
- The emerging concept of tumor and cancer cell heterogeneity also poses new challenges to our therapeutic ingenuity. In fact, tumors have been rudely awakened from their Procrustean bed and can now be seen in their naked complexity. No two tumors are alike, and arguably at some level no two tumor cells are either.
- Given the extraordinary variegated complexity of cancer and the increasing number of therapeutic armaments at our disposal, efforts are increasingly directed toward finding new biomarkers to assist clinical decision making; predicting which patients will experience the worst outcomes, selecting the best combinations of treatments, and monitoring progress during and after therapy.
- Biomarker discovery is part of a broader ambition to reclassify cancers on the basis of a molecular taxonomy in preference to the current predominantly anatomical system, which places undue emphasis on tissue of origin, visible morphology, and extent of spread.
- Why do we need a new system? Because tumor molecular phenotype and mutation status are increasingly outperforming more traditional measures when it comes to subclassification of cancers in treatment selection and outcome prediction. Although not yet true for all cancers, clinical practice in breast cancer has already been transformed with treatment selection determined by the presence of specific drug targets such as hormone receptors, HER2, and others within the tumor. Conversely, the presence of downstream activating mutations in BRAF or KRAS in colorectal cancer (CRC) identifies patients who will not respond to TKIs targeting EGFR.
- To construct this new molecular taxonomy of cancer, we have an ever expanding array of analytical techniques at our disposal. In this way, the next decade will witness the molecular subclassification of cancers with ever greater precision. However, what is the purpose of unraveling the molecular modus operandi of cancer cells unless we have new therapeutic agents to exploit it? To paraphrase Thomas Paine, “We have discovered a world of windmills and our sorrows are that there are no Quixotes to attack them.”
- Coupled with progress in non-invasive or minimally invasive techniques for sensitive detection of altered levels of proteins and various nucleic acids and in the isolation, quantification, and profiling of cancer cells from peripheral blood or other body fluids, it will become both practical and cost-effective to base future clinical decisions on the molecular biology of cancer. Taken together, one can sense that the realization of a new era of individualized medicine and tailored therapy is within our grasp.
- Ultimately, new treatments and biomarkers must be shown to be safe and clinically effective in cancer patients. Before a new treatment or diagnostic tool becomes generally available to clinicians, who wish to and can afford to prescribe it, it must first successfully negotiate a series of strictly regulated studies. Commencing with functional and toxicity studies in a number of species, safe and potentially effective agents then enter clinical trials in humans. These follow a well-defined pattern of phases, designated I–III, designed to confirm first safety and then efficacy in increasing numbers of patients. Agents progressing successfully through these trials will be afforded regulatory approval for marketing.
- Because of the expense and slow pace of clinical trials, a number of efforts are underway to find accurately predictive techniques that can suggest the optimal sequence, duration, dose scheduling, and combinations of treatments to be subsequently tested in trials. A variety of approaches are being developed, including in silico modeling, use of a range of in vitro reporter assays, and others that can determine both the PK and PD of drug activity rapidly and in such a way that meaningful predictions can be made for future activity in patients in vivo.
Introduction
“Men’s courses will foreshadow certain ends, to which, if persevered in, they must lead,” said Scrooge. “But if the courses be departed from, the ends will change. Say it is thus with what you show me!”
Charles Dickens
In one of the more surprising turnarounds in modern medical practice, cancer has for many sufferers become a chronic disease. Such a startling change in status brings new and unanticipated challenges for both carers and patients. Learning to live with cancer has, for many, taken the place of a coming to terms with the anticipated all-too-rapid death at its hands. For some cancers, exemplified by lung cancer, the outcome, though thankfully not the experience, has changed little in the last 50 years. The egregious epiphany, as one’s worst fears are solemnly confirmed in the intimidating surroundings of a clinic room, is followed in quick-fire succession by a flurry of investigations, discussions, and finally a hope-giving full-on therapeutic onslaught or a brutally deflating “sorry.” Once on treatment, matters progressed to one of three all-too-familiar outcomes, often with no time for reflection. Often in a matter of a few weeks, the patient was dead at the hands of the spreading cancer or the well-meaning clinician, or occasionally deemed to be cured. However, with all the relentless ill-fated irony of Thomas Hardy on a bad day, the celebrations would be cut short as the cancer recurred or a new one had arisen from the ashes of radiotherapy. Nowhere is this better demonstrated than in breast cancer. Fortunately, for numerous types of cancer, including breast, prostate, and colorectal, one can now anticipate a very different experience. Every day, thousands of women get up in the morning and take their breast cancers into work with them and after a normal day take them to bed with them at night – often year after year. How this volte face for cancer care came about is the subject of this chapter.
Ideally, all cancers would be cured by surgical resection and/or radiation ablation. However, such curative treatments are possible only where all of the tumor can be reached safely, and to be effective they must be deployed before cancer cells have spread. Regrettably, this is often not possible and localized or systemic radiation therapy and/or chemotherapy will be required. There are specific cases where surgery has been successfully employed to remove metastases, exemplified by local resection of solitary pulmonary metastases from colon cancer. These are however exceptional, because in most cases once cancer cells have colonized one distant tissue, they have colonized many. Moreover, even if it were possible to eradicate these established secondary deposits in some way, there will be numerous, surgically intractable cellular pioneers in transit or recent émigrés already arrived and ready to start a new secondary colony. This is not idle speculation; for many years, women underwent meretricious radical surgery for breast cancer in the vain hope that ever more drastic clearance of the breast, its surroundings, and its routes of spread might thwart the development of metastases. The blame for the failure of radical surgery can securely be attributed almost entirely to those small numbers of pioneering breast cancer cells that had slipped under the radiological and histological radar and evaded the surgeon’s knife. Thus, in some cancers seemingly localized primary tumors may already be associated with distant spread and cannot be cured by local treatments alone. For this reason, progress was dependent on the development and application of systemic treatments that could seek and destroy even the most inaccessible of cancer cells – treatments that could be deployed alone or as adjuncts to surgery.
Treatments that aim to treat or even cure cancer should by definition be more toxic for cancer cells than they are for normal cells, and the wider this disparity the better the drug. By implication, the ideal therapy will exploit unique features that separate cancer cells from their normal pristine counterparts, such as the acquired hallmark features described in Chapter 1 or the aberrant biochemical pathways and mutated molecules that gave rise to them – repetita juvant.
Interest has also surrounded the development of drugs that might arrest or contain cancer growth or spread, rather than seek to eliminate them, as these may in general be less toxic to normal cells. However, as cancer cells might not be readily rehabilitated, “capital remedies” that will leave no chance of dormant cancer cells reoffending in the future might be preferable.
Traditionally we have set at cancer with “fire and iron,” aiming to extirpate all cancer cells by surgery, radiation, and highly toxic chemicals. Most widely used chemotherapy regimens comprise drugs which exploit two basic properties of cancer cells that distinguish them (unfortunately not completely) from most normal cells: they proliferate and have aberrant DNA damage responses (DDRs) (Chapter 10). Giving drugs which interfere with the cell cycle, further interfere with DNA repair, or inflict further damage will kill cancer cells more readily than normal ones. This exquisite susceptibility to broad-spectrum agents is sometimes referred to as genotype-dependent lethality, because it is a manifestation of the entirety of the combined genotype and phenotype of the cancer cell. This is in large part the conceptual opposite of oncogene addiction and synthetic lethality, discussed in this chapter, that relate to specific individual mutations in oncogenes and tumor suppressors. Unfortunately, with traditional chemo-radiotherapy, it is rarely possible to effectively kill all the cancer cells without causing unacceptable co-lateral damage, as normal replicating cells in the bone marrow and epithelia are withered by “friendly fire.” Where sufficient normal cells can withstand the onslaught, cancers can be cured by traditional chemo-radiotherapy, notable successful examples being cis-platinum in testicular cancer and combination chemotherapy in Hodgkin lymphoma. In almost all cases, it is the inadvertent destruction of normal cells and the resultant impaired immunity, bleeding, ulceration, and sickness that set the limits on the duration and intensity of traditional chemotherapy regimens. Treatment campaigns can be designed to mitigate this by scheduling periods of cease-fire, during which time is allowed for healthy tissues, such as the bone marrow, to recover from the drug onslaught before the bombardment resumes. However, these periods of respite can also be exploited by the cancer cells to resume growth and spread. At the present time, such cease-fires are arranged for a given tumor type and anatomical grade on a one-size-fits-all basis or are individually tailored in response to comparatively crude measurements such as a full-blood count and kidney function. With the recognition that cancer cells are not all Doppelgängers has come a more nuanced understanding of tumor heterogeneity at both the molecular and behavioral levels. Cancer cell heterogeneity between patients with ostensibly similar types and grades of tumor may well lead to a new subclassification of cancers on the basis of molecular differences rather than just anatomical ones. As yet, this knowledge has had little impact on the management of most cancers, but this situation is changing rapidly.
Cancer cell heterogeneity is now known to include important differences between cells within the same patient and tumor. With this in mind, traditional agents may have limited effects on so-called dormant cancer cells that are not dividing during the treatment period, but these cells might awaken at some point in the future and thereby rekindle an apparently successfully treated cancer. The recent recognition of a small cadre of elite and particularly malign cancer cells, referred to as cancer stem cells (CSCs), poses a range of additional challenges to the ingenuity of the pharmaceutical industry. CSCs are fitted with a more resistant molecular armor and are also more securely bunkered within protected niches, making them even harder to eradicate. Yet as the perceived fons et origo of all malign behavior, they represent the perfect treatment target. Finally, as we now think of tumors as a Gesamtkunstwerk, we can also consider how the whole installation could be dismantled by cutting off the cancer cells’ support network. Thus, by subjecting the conniving stromal, vascular, and immune cells to a therapeutic onslaught, even where this has little direct effect on the cancer cells themselves, we might still bring down the entire ensemble.
So there we have the challenges, but how are we responding? It is easy to become despondent when facing the daunting complexity of tumor biology. In fact, the task of deciphering enough of it for cancer therapy to make any significant headway may seem on a par with resolving Jarndyce versus Jarndyce; no matter how many professionals earn their living in the attempt, the chance of any meaningful bequest at the end is slight. Progress has undoubtedly been made in the past, and if this owes as much to fortunate happenstance as to grand design, then we should be grateful all the same. However, the last decade has witnessed a remarkable and unequivocal shift in cancer drug development, so that with very few exceptions all new therapies are based on the fruits of unraveling the molecular complexity of cancer. The hunt is now well and truly on for drugs which, by targeting of specifically cancer-relevant proteins, have few adverse effects on normal cells. The advantages are self-evident and may extend to allowing cancer cells to be steeped in the toxic drug with minimal collateral damage. In fact, the ability to hit a cancer-critical target this hard might even be an essential prerequisite to eliminating generally treatment-resistant CSCs and dormant cancer cells.
Recent years have witnessed the addition of biologically targeted therapies to the lengthening catalogue of paradigm shifts in the natural sciences. It is uncertain that this hyperbole is truly justified as arguably we are at most simply restating Hippocrates, who suggested “contraria contrariis curantur” (the opposite is cured with the opposite) long before paradigms, epistemological or otherwise, first developed restless feet and began their now-inveterate shifting. What is, however, undeniable is that the last decade has witnessed a molecular definition of the “opposites” and, as a result, a radical reconfiguration of cancer drug development strategies. Fueled by advances in molecular biological techniques and the resultant unraveling of the molecular biology of cancer, we have largely left behind the search for nonspecific cell poisons in favor of drugs that specifically target aberrant cellular pathways – analogous to the replacement of carpet bombing everything even vaguely in the vicinity of a military target with computer and laser-guided missiles. Paul Ehrlich first introduced the term “magic bullet” in the 19th century in reference to chemicals specifically targeting microorganisms, but this idea has perfused many areas of medicine, not least oncology, in the last decade with the availability of a large and ever increasing number of new agents specifically targeting cancer cells. Of course, this strategy can succeed only if the selected targets prove accessible, essential, and irreplaceable. In the context of therapeutics, these properties far outweigh culpability, because the perfect drug target is anything that a cancer cell cannot do without and this is not always also its raison d’être. This is exemplified by the concept of synthetic lethality, whereby consequent upon the presence of various mutations, cancer cells are rendered co-dependent on noncausative genes or proteins. The appreciation of the vulnerability of cancer cells and in many cases the accurate prediction of their Achilles’ heel has proved one of the most significant victories in the battle against cancer. Like the gloved fist of a heavyweight champion, targeted therapies have ended a reputation for invincibility; cancers are no longer the implacable foe, the more perfect versions of ourselves, but are revealed as having the same glass jaw as the rest of us. Lest we forget, cancer cells had come perilously close to a transfiguration in some eyes, so it has come as something of a relief that we can now once more see them in their true guise, not the “baddest” cells on the planet, just another idol waiting for a fall. So how did we get to this turning point in the story?
From chemotherapy to targeted drugs: Chapter 15 has covered the recent era of cancer chemotherapy, so we will skim over the details here. Since nitrogen mustards first broke a lance against cancer in the 1940s and 1950s, intensive research has forged an impressive armory of anticancer therapies. Firstly, a focus on metabolic enzymes resulted in the development of drugs targeting folic acid metabolism such as methotrexate, and subsequently unraveling of the intricacies of DNA structure and replication allowed the development of agents directed against DNA polymerases and topoisomerases (Chapter 15). The first genuinely targeted agents came from advancing research into hormone signaling and the discovery that some cancer cells may still exhibit the same dependencies on hormones that characterize the cell lineages from which they were derived – this is illustrated by the effectiveness of drugs targeting nuclear hormone receptors and their ligands in breast and prostate cancer. However, it must be appreciated that such treatments will have effects on normal cells and on the organism even if these are less pronounced than with traditional DNA-damaging treatments. Thus, tamoxifen treatment for breast cancer will also self-evidently prevent pregnancy. However, one has to think laterally and monitor therapies closely. Thus, androgen depletion in prostate cancer might increase risk of and progression of type 2 diabetes and cardiovascular diseases (CVDs), though this remains contentious. If this association proves genuine, then we must consider the indirect effects of treatment on obesity and insulin levels. In practical terms, until this issue is resolved it might be wise to intensify any concurrent risk-reducing therapies for CVD in those being treated with androgen depletion.
Most recently, we have begun to really exploit progress made in unraveling the molecular biology of cancer. As discussed, all cancers are caused by a finite number of cellular genes, oncogenes, tumor suppressors, and caretakers that have been corrupted by epimutations or altered expression. The stepwise accumulation of such epimutations allows the cancer cell to proliferate abnormally and to achieve near immortality (see the multistage model for CRC in Chapter 3). Given that cancer cells also appear remarkably resourceful and will repeatedly thwart our best therapeutic efforts by mutating around them, it is all too easy to imagine some intelligent (or should that be malign?) design behind this. Yet, in reality and to paraphrase Auden, cancer cells are just “irresponsible puppets of fate or chance,” and what we are witnessing is a Darwinian process of evolution by natural selection, driven by the same forces but played out among somatic cells within the lifespan of a single organism. The essential randomness of mutations and natural selection has been underscored by rapid recent progress in molecular biology as a result of which we have now identified a truly daunting number of cancer genes, of which over 450 are thought to be causally related to the pathogenesis of different cancers. The obvious difficulty in separating next season’s hot new therapeutic targets from all those paraded on the molecular catwalk is compounded by the fact that many of the protein products on display are themselves a small part of a complex signaling outfit, comprising numerous accessory drug targets. Furthermore, many cancer-relevant products are not even on show as they are not there by dint of mutation. Moreover, it is now generally accepted that many of the mutated genes found in tumors may never have been relevant or will have become outmoded or inessential by the time treatments are administered. To borrow from Winston Churchill, this leaves our ideal drug targets “a riddle wrapped in a mystery inside a tumor,” but we also have a key at our disposal to unravel this conundrum. Thus, fortunately, we have become much better at selecting those genes and proteins that remain “mission critical” for any given cancer from this morass of potentially irrelevant passenger mutations. Step by step, we are narrowing down on a restricted and manageable range of the most tempting targets for new anticancer drugs. New concepts such as “oncogene addiction,” first coined by Bernard Weinstein in 2000, have been introduced to encompass the startling discovery that cancer cells may actually become entirely dependent on an aberrant protein, even when that same protein or pathway may be dispensable in a normal cell. The protein, to which it has become so hopelessly addicted, thus represents an “Achilles’ heel” just awaiting the inevitable sting of “Paris’ arrow” to consign the cancer cell to Homeric oblivion. Such exquisitely specific weapons not only would prove devastating to cancer cells but also should be, by comparison, innocuous to normal cells.
Targeted Therapies
Molecular differences between cancer cells and normal cells may also be exploited in other, less subtle ways. Thus, for instance, the uptake and concentration of iodine by some thyroid tumors, although shared by normal thyroid hormone-producing cells, are inconsequential in other tissues. Administering radioactive versions of iodine to patients results in the specific accumulation of radioactivity and hence restriction of damage to the thyroid, including thyroid tumor cells. In this scenario, the metabolic quirks render thyroid cancer cells vulnerable to a “targeted therapy.” More modern variants of this approach include linking toxic cargoes, such as radioactive isotopes, to antibodies or proteins, such as receptor agonists, which specifically, or at least comparatively selectively, recognize proteins expressed preferentially by cancer cells. Linking somatostatin analogues, such as octreotide, to yttrium Y 90 (a radioactive form of the metal yttrium) in order to ablate somatostatin receptor expressing neuroendocrine tumor cells is an example drawn from current clinical practice. A variety of humanized monoclonal antibodies (mAbs) are being linked to toxic cargos, particularly for use in hematological malignancies. These can be radioactive (radio-immunotherapy), in which case often mouse, rather than humanized, mAbs are preferred as they are rapidly cleared from the body. One recent example is Tositumumab used for non-Hodgkin lymphoma. Therapies that manipulate or boost the immune system to combat cancer are often grouped together as biological therapies (Chapter 13). This can create some confusion as drugs such as herceptin may be regarded as both molecular targeted agents, as they interfere with EGFR family signaling, and biological therapies as they can direct an immune attack against cells expressing HER on the surface which is bound by herceptin. Biological therapies also include a variety of cytokines including interferons and colony-stimulating factors used to counteract the negative effects of drugs on the patient’s white cell count.
Molecular Targeted Therapies
The last two decades have witnessed spectacular progress in unraveling the molecular roadmap for tumor development in many cancers. In particular, defining the central role played by kinases in growth-regulatory signaling pathways has been exploited in new drug developments. We have now entered the era of molecular targeted therapies. By exploiting the well-described differences between normal and malignant cells, new agents have been specifically developed to target gene expression and signaling pathways deregulated in the cancer cells.
So how far have we traveled along this new road? Naysayers may argue that the apparent victories of targeted therapies to date have often proved evanescent or even pyrrhic. Yet, once subjected to the spotlight of scientific scrutiny, the reasons for this, the resistance mechanisms, are being revealed. Taken together, clinical studies with targeted drugs have irrevocably altered our view of the cancer cell and have shown us how it might be vanquished. It is now hard to picture a cancer cell without superimposing a series of concentric circles and a bull’s-eye. The cure for most cancers may still appear a distant possibility, but for cancer researchers it is too late to spit out the apple. Elegantly exemplified by targeting BCR–ABL, the knowledge that, against all previous convictions, targeted therapeutics can work has proved an epiphany for cancer researchers and a clarion call to the pharmaceutical industry.
Cancer Cell Heterogeneity
The application of extraordinarily powerful analytical tools for defining cell phenotype supports a view that you may feel is self-evident, namely, that no two cells are exactly alike. They may share a common ancestry and location, and even have several mutations in common, but invariably at the level of gene or protein expression there will be differences, and these differences will be reflected in different behavior and function. Many cancers, exemplified by breast cancer, are now known to represent a far more heterogeneous group than was previously appreciated (intertumoral heterogeneity). Thus, seemingly similar cancers may in different individuals be associated with widely differing clinical features, natural history, drug sensitivities, and prognosis. In part, such differences may reflect the different cellular origins of the cancer. Thus, stem cells or more differentiated cells will start at different points and follow a different roadmap to malignancy. In fact, it appears that even terminally differentiated cells can be encouraged back into the cell cycle. Stem cells may more readily be able to propagate newly acquired mutations, whereas differentiated cells would first have to forsake their vows. However, auspicious mutations can reawaken a latent reproductive appetite in even the most G0-entrenched celibate. Subclassification of cancers, often described as grading or staging, has long been employed by oncologists to assist decision making. However, despite some improved codification, this process is still largely based on gross radiological and histological appearances, which may miss quite substantial and clinically vital differences at a molecular level. Biomarkers are discussed elsewhere, but it suffices to say that cancers can be further subdivided on the basis of molecular phenotype.
Once we examine cancer at a detailed molecular level, it rapidly becomes apparent that not only are no two tumors quite alike but also, probably, no two parts of the same tumor are (intratumoral heterogeneity). Moreover, the cells within a given single tumor show extensive variation. Not only do tumors comprise cancer cells, CSCs, stromal cells, endothelial cells, and immune cells, but also each of these different lineages encompasses cells of differing states of differentiation and clonal origin. Moreover, essentially all of these will show considerable dynamic variation in gene and protein expression over time. Recently, the theory, developed by Joan Massague and colleagues, that primary tumors may be self-seeded or reseeded by circulating cancer cells provides a further driver of cell heterogeneity. In fact, as our tools allow an ever more comprehensive survey of gene and protein expression, so the immense complexity as well as the unexpected heterogeneity of the cancer genome between and even within apparently histologically identical tumors are revealed. This does not automatically imply that earlier anatomical taxonomies of cancer are valueless. This is far from true, but we must acknowledge that they are imperfect and in some cancers unable to predict prognosis with even modest accuracy. In some cases, molecular phenotype and histology may be closely associated, implying that abnormalities in given signaling pathways may result in a reproducible observable phenotype.
Histological or Molecular Classification
In non-small-cell lung cancer (NSCLC), therapy decisions can be based on the gross histology (squamous vs. adenocarcinoma) and clinical parameters (smoking status, ethnic origin, and age). Particular mutations again show some relationship to these more traditional classifiers, which can be employed to direct targeted treatments more appropriately. Thus, squamous cell tumors respond poorly to first-line EGFR inhibitors due, for example, to the presence of downstream activating mutations in RAS. Thus, despite their limitations, currently employed analytical techniques may still serve practically until we can replace them with something better. However, histology will not identify the point at which signaling is aberrant and can be misleading where observable changes in phenotype are nonspecific. Therefore, if histology cannot be relied on to make sufficiently accurate treatment choices, then ultimately all cancers will have to be subjected to a molecular analysis in order to tailor treatments appropriately. This is exemplified by traditional histological categories of invasive breast cancer, such as luminal A, luminal B, and basal-like, which are often linked to specific mutations, but as these are not sufficiently reliable, it has already become commonplace to look for the presence of specific molecular changes, such as receptor status, before selecting treatments (discussed further in this chapter).
Molecular Classification in Breast Cancer
This clinically heterogeneous cancer is currently classified into three broad molecular categories:
- Estrogen receptor (ER) and/or progesterone receptor (PR) positive tumors responsive to anti-hormone treatments.
- HER2 (ERBB2) amplified, responsive to mAb and TKIs directed against HER2.
- Triple-negative breast cancers (lacking expression of ER, PR, and HER2), with increased frequency of mutations in BRAC1. These tumors are also known as basal-like breast cancers and are currently treated with chemotherapy options, though PARP inhibitors may offer a targeted treatment.
Recent studies have increasingly focused on more complex molecular analyses, and expression of a variety of other pathways appears to be helpful in prognosis and may identify future targets. These include TP53, PIK3CA, and GATA3, common to multiple histological subtypes, and specific mutations in GATA3, PIK3CA, and MAP3K1 found in luminal A.
Future Directions in Molecular Classification
This area is advancing rapidly, and it will soon be commonplace to look for key oncogenic drivers in NSCLC, such as amplification of TKRs, mutant KRAS, or EML–ALK fusions. Although still a very simplistic molecular phenotype, classification on the basis of presence of these mutations appears to identify mutually exclusive groups of tumors (though note recent studies in glioblastoma, discussed in this chapter, and the problem of intratumoral heterogeneity). Moreover, whatever the theoretical limitations, there is practical value where molecular classification is gainfully directed at targets for available and emerging treatments. But what of known mutations in other oncogenes, in tumor suppressors and differentiation markers, and in gene–protein expression and also in cellular heterogeneity?
Is there a clinical point to an ever more divergent subclassification of individual tumors based on an ever more complete description at a molecular level? Is it not sufficient to catalogue tumors on the basis of what treatments will eradicate them? In some cases this may suffice, and knowing that most cancer cells have mutations in a growth factor TKI or have lost a given tumor suppressor can be exploited in selection of a particular therapy. Put simply, subdividing cancers hierarchically on the basis of shared anatomical or molecular features may enable ever improving accuracy of clinical decision making, but in some cases the added gain in moving from broader, less comprehensive, but probably cheaper levels down to ever narrower subclasses based on detailed molecular analyses may be unnecessary. However, in most cases it is accepted that improved molecular subclassification of cancers is urgently needed. For example, at present assessment of prognosis in the intermediate stages of CRC is very imprecise, with widely varying 5-year survivals within apparently identical grades of tumor.
Revealing Heterogeneity Across Single Cells
Averaging expression or genome analyses across multiple cells may fail to reveal subtle differences between individual cells, including the existence of cancer stem cells, rare cells with additional mutations that might identify individuals likely to rapidly become resistant to particular targeted drugs, and so on. Moreover, where the population of cells is in the minority, their contribution may be masked.
What causes such heterogeneity? At a superficial level, the inherent diversity of biological systems is expanded during tumorigenesis by mutations, clonal evolution, and genomic instability. On top of these, we must also factor in epigenetic changes, ongoing differentiation along diverging lineages, and more transient effects of cell plasticity. Thus, interactions with the microenvironment, or niche (comprising other cancer cells; noncancer cells, including stroma, endothelium, and immunocytes; and those with cell-derived proteins and biophysical features such as low oxygen tensions), can all influence molecular phenotype and even encourage the expression of stem cell–like characteristics. Even cancer stem cells may display extensive plasticity that may allow them to alternate between cancer-expanding epithelial and metastatic or invasive epithelial–mesenchymal transition (EMT) phenotypes depending on their microenvironment, differentiation state, and metabolic parameters, including oxygenation and glucose metabolic pathways.
Thus, accurate analyses of cancers may be achieved only by analyzing single cells, if possible within their anatomical and spatial contexts (see Chapter 20). Improved understanding of the drivers behind tumor cell heterogeneity as well as of the resultant functional sequelae of diverse cell–cell interactions will prove an essential prerequisite to developing new treatment strategies for cancer. This follows from the desire to eliminate all cancer cells. Thus, individualized therapies may need to be tailored not just to the individual patient and tumor but also to its many varied and continually evolving cellular constituents.
Cancer Heterogeneity and Therapy
In fact, intratumoral heterogeneity may pose a substantial barrier to molecular targeted treatments, because their success will depend on the molecular and cellular diversity in individual tumors. This can be problematic for numerous reasons, and a few are suggested here. Not knowing what mutations are or may become present in other cancer cells in many cases could prevent us from thwarting the emergence of resistance by using a strategic combination of drugs. This view is receiving strong support from the improvements in outcome in breast cancer resulting directly from molecular phenotyping and the ability to select appropriate therapies based on that information. Furthermore, in glioblastoma several studies have now confirmed that driver mutations long known to coexist within tumors and contribute to progression are rarely present in the same tumor regions, let alone cells. Thus, EGFR, MET, and PDGFRA may all be amplified in a single tumor, but in a mutually exclusive pattern across differing single cells. Such cohabitation of multiple different clones, carrying activation of different pathways, within the same tumor, even where many may share a common ancestry, will affect the choice of targeted therapies and may predict the likelihood of resistance.
Finding the Molecular Targets
Let us now consider how we select biological drug targets from among the many potential candidates. Broadly, we must progress from a descriptive to a functional understanding of cancer cells by the application of a range of research techniques grouped together as “functional genomics,” the executive arm of systems biology (Chapter 20). Why? Put simply, because identifying missing, overexpressed, or altered proteins in cancer cells does not necessarily mean they are mission critical for oncogenesis or would represent good treatment targets. Thus, by using combinations of cultured cell lines, genetically altered mouse cancer models, and implantation of human cancer cells into suitable recipient models, we can work out what all the myriad genes, proteins, and their mutant variants actually do, with whom, and for what results. This is no mean task, given the heroic numbers of potentially interacting protein partners. Of course, this pipeline can only suggest potentially druggable targets. Ultimately, these will need to be fully validated in clinical trials, but such functional studies can greatly reduce the numbers of targets that need to be explored in this way, with all the potential cost savings in financial and human terms. Other chapters discuss some of the methodologies employed to uncover the functional roles played by cancer-relevant molecules and to predict which may be the most optimum new drug targets (Chapters 6 and 20); next, we will specifically look at the all-important surrogates for testing the potential of targeting specific genes and proteins in vivo – conditional genetically altered organisms.
Tumor Regression in Mice by Inactivating Single Oncogenes
What sense would it make or what would it benefit a physician if he discovered the origin of the diseases but could not cure or alleviate them?
Paracelsus
There is little doubt that functional studies offer the best hope of separating wood from trees and the propitious mutations from the capricious. New strategies targeting oncogenes (or other cancer-contributing mutations) can be investigated by employing model systems, even before a suitable drug has been developed. The use of mouse models in oncogene research follows on from two postulates: firstly, that mouse models provide valuable insights into how oncogenes work, particularly when combined with studies of human cancers; and, secondly, that they allow therapeutic strategies, including timing and dosing schedules, for oncogene-targeted agents to be evaluated.
Compelling results from rodent models and cell culture experiments suggest that several oncoproteins, including MYC, RAS, and RAS-signaling pathways, such as the RAF–MAPK and PI3K pathways, might prove effective treatment targets in cancer, with the important caveat that successful results in treating human diseases will have to be confirmed. Intriguing paradoxes have been revealed by several studies; thus, for example, the demonstration that transient inactivation of MYC may be sufficient to arrest and reverse certain tumor models, but not others, raises the importance of tissue microenvironment and epigenetic context in dictating the potential therapeutic responses of any given tumor.
With the emergence of conditional transgenic mouse models, in which the expression or activation of a given gene or protein could be regulated (Chapter 6), has come a new and more functional understanding of the role played by cancer mutations in cancer biology. The study of such models has resulted in the overturning of a number of articles of faith pertaining to the likely benefits of targeting single molecules in cancer. Thus, it was generally assumed that several oncogenic pathways would need to be targeted for tumor regression to occur. Surprisingly, this turned out not to be the case, and targeting only one specific gene or protein in a cancer often leads to tumor regression. The concept of “oncogene addiction” has gradually effloresced from such studies (discussed in this chapter); broadly, the consequences are that the survival of a cancer cell depends on the continued activation of the particular oncogene, such as MYC or RAS – inactivating the oncogene leads to death or senescence of cancer cells (Fig. 16.1). Of course, it is important to remember that although oncogene inactivation can cause tumor cell death at one particular stage of cancer, most tumors eventually recur, and we need to understand when and why this occurs in order to design therapeutic strategies to treat cancer (Fig. 16.1). Here, we will highlight some of the mouse models of cancer (Table 16.1) that have led to the discovery of “oncogene addiction” as well as cases in which tumor relapse occurs. It is also important to understand that animal models, along with cultured cancer cells, can provide unique opportunities to test the functional importance of a given cancer protein or gene without having to first develop a suitable drug. This is critical as it is often difficult to identify which of the many mutant genes or proteins in a cancer are actually “mission critical” – in other words, to separate the propitious from the capricious.
Table 16.1 Use of regulatable transgenic mouse models of cancer to test tumor regression
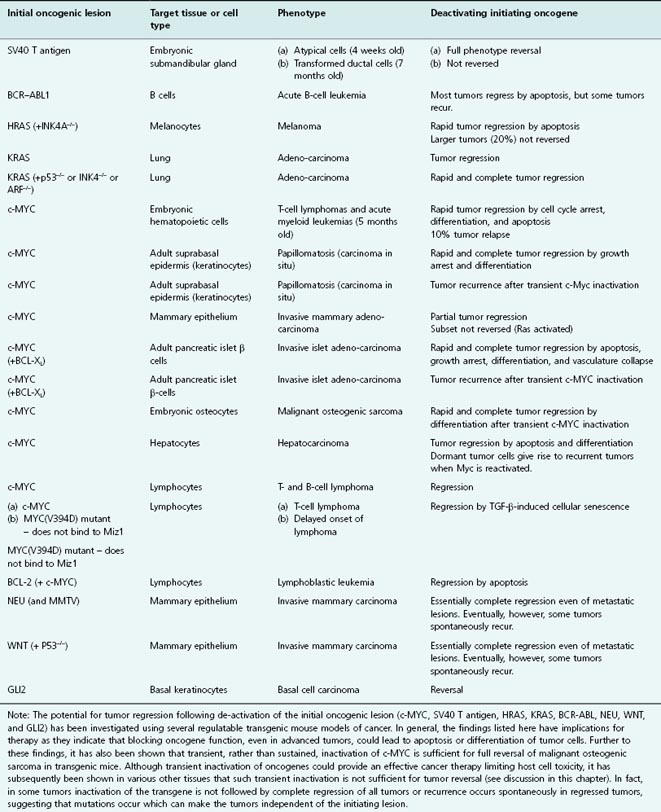
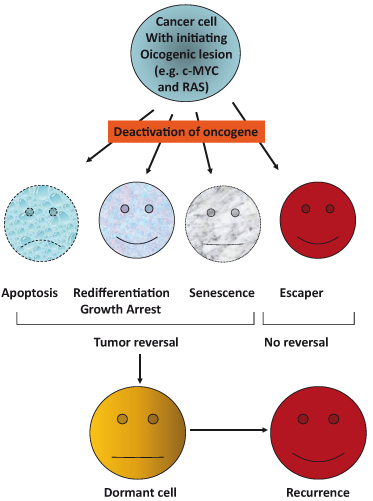
Figure 16.1 Tumor reversal following deactivation of initiating oncogenic lesion. Studies in numerous different transgenic mouse models of tumorigenesis have revealed that neoplastic lesions can be completely and often rapidly reversed when the initial causative oncogenic lesion is inactivated. Mechanisms of tumor regression include tumor cell apoptosis, growth arrest concomitant with differentiation and re-establishment of cell–cell contacts, and/or cellular senescence. The rapid onset of collapse of the vasculature, within tumor masses, may also contribute to tumor cell apoptosis. However, in some cases, a subset of tumors escapes reversal (“escapers”) because they are no longer dependent on the initiating oncogenic lesion. In these cases, it is likely that additional oncogenic lesions have rendered the initial lesion redundant. In addition, cancer cells may avoid elimination by effectively lying dormant for a period of time before becoming reactivated in some way and resuming their oncogenic mission.
As was mentioned for MYC in Chapter 6, in general two major approaches have been used to generate conditional (regulatable) transgenic mouse models: the tetracycline (“tet”) system and the modified estrogen receptor ERTAM (see also Appendix 20.1). The tet system requires the drug doxycycline to regulate expression of the gene of interest, whilst the ERTAM system relies on the administration of tamoxifen to regulate activity of the expressed protein (see Fig. 6.9).
The key advantage of these systems is that a given gene or gene product can be switched on in a specific tissue in a time-controlled manner, which allows the close monitoring of tumor development. Conversely, the gene–gene product can subsequently be switched off once tumors have developed, providing insight into mechanisms of tumor regression.
What we can derive from these studies is that inactivating the initiating oncogene (e.g. MYC or RAS), in tumors often results in tumor regression as a result of proliferative arrest, differentiation, senescence, and/or apoptosis. The outcome is dependent on the genetic alterations as well as the tumor or tissue type.
Conditional Models Systems to Study C-MYC in Cancer Biology
Using the tet system, mice were generated that conditionally express MYC in their hematopoeitic cells. In this mouse model, MYC was expressed in this cell lineage throughout development, resulting in the occurrence of highly invasive T-cell lymphomas and acute myeloid leukemias by 5 months of age. Remarkably, subsequent inactivation of the MYC transgene was sufficient to cause tumor regression in mice moribund with tumor burden; tumor burden was substantially reduced within 3 days, and after 6 weeks the majority of mice exhibited a sustained remission for as long as 30 weeks. Downregulation of MYC expression resulted in both the elimination of tumor cells by apoptosis and the differentiation of others into mature lymphocytes with the restoration of normal host hematopoiesis. However, despite sustained tumor regression in the majority, 10% of mice develop recurrent tumors. Thus, some cancer cells in regressing tumors escape dependence upon the MYC transgene. Such “escapers” have probably acquired genetic lesions that substitute for the requirement for MYC and will repopulate the tumors (Chapter 6). In another cancer model, in which both MYC and the antiapoptotic protein BCL-2 are expressed in lymphocytes, switching off BCL-2 leads to complete reversal of lymphoblastic leukemia by apoptosis. Further examples are given in Table 16.1. The central importance of interactions between Myc and Miz1 in initiating and sustaining T-cell lymphomas has been demonstrated in a tet-regulatable mouse model, described in Chapter 6; reversing Myc expression restores the expression of growth-inhibiting genes, such as those for cdkn2b and cdkn1c, that were previously suppressed by Myc–Miz1 complexes leading to senescence. Tumor-derived TGF-β may contribute to this.
Regression of several different solid tumor types has also been shown, following MYC deactivation in regulatable mouse models, including skin papillomas, pancreatic islet β-cell, osteosarcoma, breast adenocarcinoma, and hepatocellular carcinoma. As described in Chapter 6 (see Fig. 6.9), continuous activation of MYC (using the c-MYCERTAM system) in skin epidermis led to the growth of benign, angiogenic papillomas. Inactivating MYC led to the complete regression of papillomas over a 3-week period which coincided with skin keratinocytes exiting the cell cycle and resuming normal differentiation. Importantly, the complete regression of newly formed vasculature demonstrated that continuous MYC activation in keratinocytes is required to maintain neo-angiogenesis.
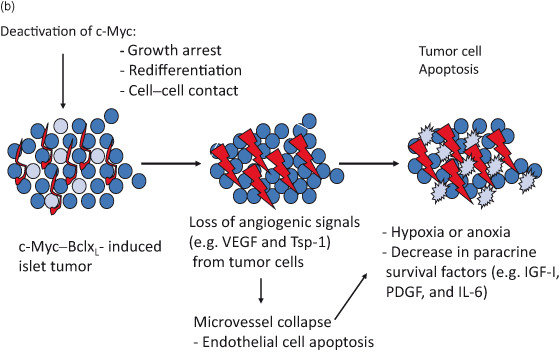
In a pancreatic islet β-cell tumor model, multiple angiogenic and locally invasive β-cell tumors, which develop following continuous activation of MYC and the antiapoptotic protein BCL-XL (described in Chapter 6 and Fig. 6.10), were also completely dependent upon active MYC. In this tumor model, regression appears to be mediated by a rapid collapse of vasculature that triggers the death of many β cells, which is presumably the result of hypoxia, lack of nutrients, and survival factors provided by the vasculature. Remarkably, despite the continued overexpression of BCL-XL in neoplastic β cells, deactivating MYC leads to general regression of tumors back to fully differentiated quiescent islets with restoration of normal cell–cell contacts concomitant with re-expression of E-cadherin (Fig. 16.2). Interestingly, tumor regression is also accompanied by substantial infiltration of inflammatory cells, which may play a role in phagocytosis and the clearance of apoptotic and necrotic debris. Recent studies suggest that interactions between activated oncogenes and immune cells may play a role in shielding the cancer from immune attack – a protection which may be lost when oncogenes are inactivated. The rapid disassembly of vasculature is consistent with a direct angiogenic role of MYC, described in this chapter, although it is also possible that β-cell growth arrest induced by MYC inactivation allows intrinsic anti-neoplastic mechanisms such as the immune response to initiate regression. With regard to ectopic β cells that had invaded pancreatic ducts, vessels, and local lymph nodes by 12 weeks of MYC activation, extensive histological analysis of pancreatic sections failed to reveal any ectopic β cells after several weeks of MYC inactivation. Moreover, other animals undergoing similar regression remained healthy after several months. It is not yet known, though, if longer term activation of MYC would lead to a progressively increasing proportion of tumors escaping dependence upon MYC, as observed in other tumor models.
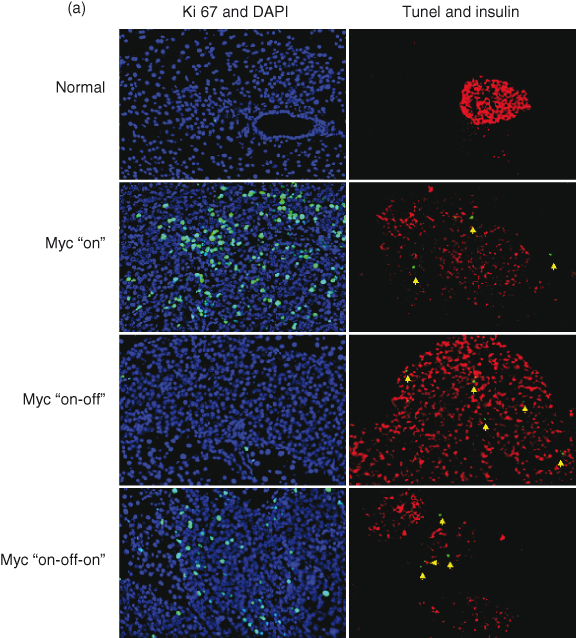
Figure 16.2 Reversal of murine pancreatic islet β-cell tumors. (a) Deactivation of c-MYC in pancreatic islet tumors results in complete reversal in the overwhelming majority of tumors by 4 weeks after deactivation. Here we show the effects of transient inactivation of c-MYC. After a period of 5 days of inactivation, β cells have stopped replicating (absence of Ki67 staining: green) and have acquired a more differentiated phenotype (enhanced expression of insulin: red). Moreover, some β cells are undergoing apoptosis during the period of c-MYC inactivation (TUNEL positive: green). Those cells undergoing apoptosis whilst c-MYC is on are not β cells as they do not express β-cell markers. (b) The processes of tumor reversal are shown schematically.
An interesting study has shown that brief inactivation (10 days) of MYC is sufficient for the sustained regression of MYC-induced invasive osteogenic sarcomas in transgenic mice. Surprisingly, subsequent reactivation of MYC led to a “reversal of fortune” for the cancer cells as these now undergo extensive apoptosis rather than restoration of the neoplastic phenotype; for some reason, MYC had crossed the house and was now a committed anticancer representative. One possible explanation for this outcome is that a change in maturity has taken place. While MYC was inactivated, the epigenetic context has shifted and the cell is no longer the immature one in which MYC was originally activated, but rather a more differentiated one with a resultant different response to MYC activation; the immature osteoblasts present during embryogenesis, as well as the osteosarcoma cells deriving from them, are very different from the mature osteocytes into which they differentiate during the period of MYC deactivation, and respond very differently to MYC. Therefore, reactivation of MYC now takes place in a different cellular context and induces apoptosis rather than neoplastic progression.
These intriguing findings suggested the novel possibility of employing transient inactivation of MYC as a therapeutic strategy in certain cancers, thus limiting potential toxic effects that result from prolonged therapeutic inactivation. But how general are these findings, and what happens in other tumor types? Unfortunately, brief inactivation of MYC failed to reverse tumor growth in several different mouse models. In contrast to the osteogenic sarcoma model described in this chapter, reactivating MYC in islet tumors does not lead to accelerated β-cell apoptosis, but rather restores the oncogenic properties of MYC, rapidly re-initiating β-cell proliferation, loss of differentiation, local invasion, and angiogenesis. In epithelial tumors, such as liver and breast cancer, reactivation of MYC in regressed tumors also led to tumor regrowth. Importantly, subsequent inactivation of MYC was less effective at regressing tumors, indicating that other oncogenic pathways are being activated in these cells.
In mice that had developed hepatocellular carcinoma, inactivating MYC promoted apoptosis as well as terminal differentiation of many tumor cells, giving rise to “normal” liver cells – hepatocytes and biliary cells. However, although such cells appeared and behaved as “normal,” some indeed retained oncogenic properties, as was seen following MYC reactivation. So the important message here is that sustained inactivation of MYC is required for tumor regression, as some “normal” tumor cells are actually in a state of dormancy and upon reactivation of MYC can unleash their neoplastic behavior.
An important point to remember is that different types of cancers are prevalent in different age groups with the effects of oncogene activation dependent on the developmental stage of the target cell at that time. Thus, the biological consequences of activating oncogenes such as MYC are clearly influenced not only by environment but also by developmental stage. This has been elegantly shown in two mouse models, mammary gland and liver. MYC can inhibit postpartum lactation if activated within a specific 72-hour window during midpregnancy, whereas MYC activation either prior to or following this 72-hour window does not. In embryonic and neonatal mice, MYC overexpression in the liver immediately results in hyperproliferation and neoplasia, whereas in adult mice MYC overexpression induces cell growth and DNA replication but without mitotic cell division, and neoplasia is considerably delayed.
Taken together, these findings suggest that a cautious approach is required in considering cancer therapies aimed at brief, as distinct from sustained, oncogene inactivation. Firstly, a more comprehensive understanding of the genetic basis and environmental context of any individual tumor would be required in order to predict the likely success of such a treatment schedule. Secondly, at least under those circumstances where tumor cell differentiation and alteration of epigenetic context would not be predicted to reinstate apoptosis and no alternative mechanism exists for tumor cell removal, sustained inactivation of the offending oncogene would seem the desired therapeutic goal.
Conditional Models Systems to Study RAS in Cancer Biology
Well, what of RAS? Much of the methodology employed has been discussed, so we will restrict ourselves to a brief overview of transgenic studies here. In a mouse model of melanoma, withdrawal of doxycycline-inducible oncogenic HRAS(G12V) expression in transgenic mice bearing melanomas causes apoptosis in both tumor cells and endothelial cells, followed by regression of the melanomas. Similarly, withdrawal of doxycycline-inducible activated KRAS(12D) expression from type II pneumocytes caused apoptosis and regression of both early proliferative lesions and lung cancers. Similar results were obtained in animals deficient in p53 or p19Arf. Thus, activated HRAS or KRAS is required for tumor maintenance in mouse models of melanoma and NSCLC, respectively.
RAS and MYC Together
Several studies have now looked at oncogene deactivation in mouse tumors related to combined deregulation of both MYC and RAS. Genetic context is clearly important in dictating the consequences of inactivating MYC – so what happens if RAS is also deregulated?
A well-established mouse model of NSCLC was used (LSL–KrasG12D) in which activation of the transgene K–RasG12D is initiated in mice by inhalation of adenovirus expressing Cre recombinase (for technical details on how this works, see Chapter 20). Multifocal lung tumorigenesis then develops, and by 18 weeks each lung harbors multiple independent tumors at all stages of evolution through to adenocarcinoma. The idea was to cross-breed Omomyc mice with LSL–KrasG12D mice in order to study both the therapeutic impact of blocking MYC activity in lung tumors as well as the side effects of inhibiting MYC in normal cells.
Remarkably, in as little as 3 days Omomyc expression triggered profound tumor shrinkage and, after 28 days, animals were overtly tumor free. With regard to the impact of inhibiting MYC systemically in normal cells, surprisingly, only mild and well-tolerated side effects were evident in normal proliferating tissues.
Mice showed no signs of distress; maintained their weight, hydration, and normal blood chemistry; and, while proliferating tissues such as intestine, bone marrow, skin, and testis exhibit varying degrees of attrition, cell death does not occur in any adult tissue, all of which maintain structural integrity. Importantly, all effects of MYC inhibition on normal tissue are fully and rapidly reversed upon restoration of endogenous MYC function.
These surprising observations strongly support rekindling interest in MYC as a therapeutic cancer target for two obvious reasons: firstly, endogenous MYC is required not only for the proliferation of RAS-driven lung tumors but also for their survival (inhibiting MYC with Omomyc leads to tumor regression); and, secondly, such dependency on endogenous Myc for cell survival is specific to tumor cells and absent from all normal proliferating cells.
In general, these findings have implications for therapy as they indicate that blocking oncogene function, even in advanced tumors, could lead to apoptosis, senescence, or re-differentiation of tumor cells (Fig. 16.1). The demonstration of tumor regression following deactivation of initial oncogenic lesions in mouse models (e.g. MYC, SV40 T antigen, RAS, BCR–ABL, and BCL-2) has provided valuable information and hope for future development of candidate drug molecules. To this end, recent in vivo studies show that re-engaging apoptosis pathways, which have become disrupted during tumor development, can indeed have a positive therapeutic effect. For instance, the inhibition of the antiapoptotic protein, BCL-2, or the restoration of p53 function has proven particularly lethal to particular tumor types. Even if targeting the oncogenes themselves may not be a viable strategy, various downstream targets mediating oncogenesis might. Over the last few years, a large number of studies have employed gene chip microarrays, deep sequencing, and other high-throughput techniques to identify oncogene-regulated genes in cultured cells in vitro. However, all these studies, despite their undoubted utility, do not give us any direct indication of which of these many oncogene-regulated genes are actually involved in tumorigenesis in vivo. Yet this is of great importance given the critical role of the tumor microenvironment as well as factors operating at the organism level. Taken together, these findings support the notion that a detailed understanding of the “road map to cancer” of a given individual tumor may be the essential prerequisite to selecting optimal therapeutic strategies in the future, thus providing some impetus for strategies aiming to achieve one of the great hopes of postgenome-era biology, namely, “individualized medicine” and “tailored therapy.”
Resistance and Recurrence in Mouse Models
As mentioned, there are some instances when tumors do not fully regress (Table 16.1). Although it is not clear as to why some tumors escape dependence upon the initiating oncogene, it has been reasoned that these tumors might have acquired further genetic lesions that in some way substitute for the requirement of the oncogene.
For example, in a mouse model of breast cancer, although reversal of MYC-induced invasive mammary adenocarcinomas occurred, a subset of tumors failed to reverse and was subsequently found to carry additional mutations in RAS. The observation that MYC may cause genomic instability in some model systems in vivo and in vitro might in the long term contribute to MYC-induced neoplastic progression. Further examples of mouse models in which tumors subsequently relapse are given in Table 16.1. Other specific examples are discussed in more detail later in the chapter in the sections on resistance to targeted treatments.
Targeted Cancer Therapies
A desire to take medicine is, perhaps, the great feature which distinguishes man from other animals.
Sir William Osler
The modern era of cancer therapeutics is increasingly based upon the exploitation of new knowledge deriving from the unraveling of cancer cell biology, supported by functional studies in surrogate models. Targeted cancer therapies that block the growth and spread of cancer by interfering with specific molecules involved in tumorigenesis and tumor growth effectively antagonize the hallmark features of cancer. Such agents are also known as “molecular targeted drugs.” By targeting molecular and cellular changes that are specific to cancer, targeted cancer therapies may be more effective than current treatments and less harmful to normal cells. Although cancers are remarkably heterogeneous, they share certain key properties. Thus, all cancer cells exhibit uncontrolled and excessive replication; are generally very long lived; and, not surprisingly, as well-differentiated cells rarely if ever divide, are less differentiated than their normal counterparts. This is emphasized by the two broad functional categories into which new targeted agents in cancer may be separated: differentiation therapies (cytostatic) or destruction therapies (cytotoxic). Given that new therapies which simply arrest growth of the primary or act to prevent invasion or metastasis will not result in major radiographically apparent shrinkage or ablation of the tumor, therapeutic evolution will need to be matched by equally rapid progress in new measures of treatment success to run alongside more conventional measures of tumor volume, such as Response Evaluation Criteria in Solid Tumors (RECIST).
Targeting Oncogenes to Treat Cancer?
Sane judgment abhors nothing so much as a picture perpetrated with no technical knowledge, although with plenty of care and diligence.
Albrecht Dürer
The recent appreciation that cancer cells may actually become critically dependent on an aberrant active oncogene (oncogene addiction; see Fig. 16.3) raises the possibility of developing agents specifically blocking the activity of these mutated or deregulated oncogenes with few harmful effects on normal cells. Moreover, as by definition oncogene-addicted cancer cells must have also acquired resistance to the intrinsic tumor suppressor action, apoptosis or senescence, invoked by deregulated oncoproteins, such cancer cells would also be vulnerable to the therapeutic reactivation of the interdependent inhibitory pathway. In fact, cancer cells may be very vulnerable to restoration of a missing or failing tumor suppressor, and this is sometimes referred to as tumor suppressor hypersensitivity, analogous to oncogene addiction. Finally, cancer cells may also become critically dependent on normal cellular processes that are redundant or supernumerary in normal cells; described as non-oncogene addiction, use is restricted to genes that are not themselves mutated in cancers. Such genes are disproportionately important to cancer cells, which have been dispossessed of otherwise interchangeable alternative pathways needed to complete some essential process. The exemplar par excellence of non-oncogene addiction is the complete reliance of BRCA-mutated cancer cells on an alternative DNA repair pathway involving PARP; such BRCA mutant cells are, therefore, ill equipped and lack the resources required to endure treatment with PARP inhibitors.
Figure 16.3 Oncogene addiction. The addicted cell suppresses alternative growth-stimulating pathways, making it more vulnerable than a normal cell to drugs blocking the addicted pathway.
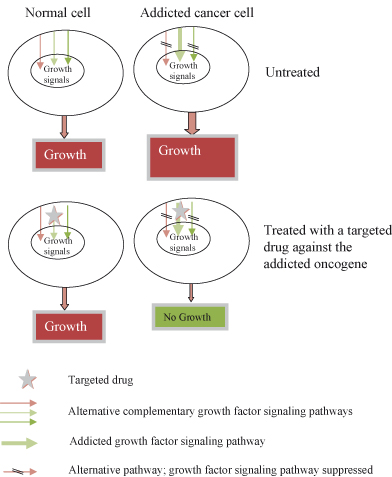
Identifying critical interdependent signaling pathways as therapeutic targets in mature cancer cells is a closely allied goal to the unraveling of the “cancer road map” that guided their development in the first place. However, with a critical difference that has considerable practical relevance, mutations that may have been essential in primordial cancer cells might no longer be at the point in time that treatments are mooted. This issue can be resolved only by functional studies in which the consequences of inactivating a cancer-causing target can be examined directly; simply describing whether a gene is mutated or not is insufficient, as in this particular cancer and at this particular time that gene may have ceased to be relevant. Progress has been made in this regard, and over the last few years numerous functional studies in animal models have confirmed that many cancers can be successfully reversed by inactivating the initiating oncogenes, such as c-MYC, KRAS, HRAS, and BCR–ABL, or by restoring the function of missing tumor suppressors, such as p53, that have been neutered by cancer-causing epimutations. The big surprise here is the dearth of failures in such studies that often demonstrate meaningful effects with small numbers of replicates, which seems in direct contrast to drug trials in humans. In part, this might reflect the pernicious influence of publication bias; negative studies are deemed boring and are often rejected out of hand or are relegated to seldom-consulted publications, but this applies even more strongly to drug trials in cancer patients. More likely, the remarkable genetic homogeneity of animal models encourages reproducibility, whereas the great diversity of outbred animals including humans largely ensures the opposite, necessitating large numbers of study participants to achieve significance.
Despite these shortcomings, the study of model systems has revealed several interdependent oncogenic signaling pathways (as well as the tumor suppressor pathways engaged by them), which must be overcome for tumorigenesis to proceed. Indeed, when oncogenes are deregulated without such other cooperating lesions already in place, the cells die or senesce rather than proliferate. Most cells are remarkably ambivalent about their own survival and will readily embrace apoptosis when this is mandated or scheduled. Although for some time believed to be a misconception related to differences between animal models and humans, we now know that under some circumstances cancers can develop through the conflation of as few as two oncogenic signaling pathways even if seven or more may be the norm. This is encouraging, as it supports the emerging view that cancer cells, despite their increasingly complex molecular divergence from their forbears, may at heart remain vulnerable to targeting of these primordial founding lesions. Proof-of-concept studies in humans for targeting the “dastardly duo” of MYC and RAS must await the arrival of suitable therapeutics. However, the notable successes in clinical trials of a number of TKIs directed at BCR–ABL and EGFR as well as antibodies directed at HER2 and of a variety of others in various stages of clinical trials affirm that even well-established cancers are still in a state of original sin, and might be redeemed and restored to a prelapsarian purity by curing them of their originating molecular corruptions.
Tumor cells become addicted to mutations that activate growth-regulatory signaling pathways, possibly due to the resultant activation of compensatory inhibitory feedback loops that effectively repress all other growth-promoting pathways. Thus, cancer cells with mutations in growth-regulatory pathways are much more sensitive to specific drugs that inhibit those pathways than are cells in which these pathways are activated by physiological means (unless, of course, the drugs target the negative feedback loop as well; see the “Resistance to targeted therapies” section). However, each time the cancer cell loses a growth-promoting pathway to a new therapy, the mutational wheel of fate spins again and another deregulated growth pathway may take its place. These resistance pathways will be discussed in this chapter.
Targeted cancer therapies are being studied for use alone and in combination with each other and traditional chemotherapies or as adjuvants in combination with surgery or radiation (Chapter 18 discusses those that are in clinical use at the present time). Most of these new drugs target proteins that are involved in growth-signaling processes, in particular, though not exclusively, the tyrosine kinases. One great hope is that more detailed identification of the molecular phenotype of individual patients and their tumors will ultimately bring in the new age of individualized medicine with treatments ultimately “tailored” to fit the unique set of molecular targets present within any given individual patient’s tumor. Targeted cancer therapies also hold the promise of being more selective, thus minimizing collateral damage to normal cells, improving tolerability, and improving quality of life.
These specific agents will be discussed in more detail in this chapter and in Chapters 15 and 18.
Oncogene Addiction
You are invulnerable, you have no Achilles’ heel.
You will go on, and when you have prevailed
You can say: at this point many a one has failed.
T.S. Eliot
The term “oncogene addiction” has been used to encompass a range of potential mechanisms whereby a cancer cell becomes critically dependent on a given activated oncogene for survival and/or proliferation (Fig. 16.3). This has evident implications for the potential success of targeted cancer treatments. Yet, if the number and complexity of targets are beyond our understanding, then will this hard-won knowledge be of any value? Well, there are good reasons for optimism. Firstly, despite the seemingly bewildering array of mutations, epigenetic alterations, and differential expression of genes and proteins between cancer cells and their normal forebears, cancer cell behaviors are much more limited. In fact, the essential banality of cancer is illustrated by how readily behaviors can be distilled into a short list of shared hallmark features. However, the limited repertoire belies the variety of molecular contributors; those directly involved approached 500 at last count.
Recent studies have begun to unravel the molecular basis behind oncogene addiction, and some common themes have emerged. Thus, the targeting of proteins to which cancer cells are addicted will have far greater impact on their growth than on that of normal cells, allowing the use of higher doses with fewer side effects. One can also target the ancillary factors that enable the addicted oncogene to direct aberrant growth. Thus, mutations in MYC and RAS, to name but a few, are actively oncogenic only if they are uncoupled from pathways regulating apoptosis and senescence; re-engaging these processes will allow cancer cells to kill themselves or to renounce replication. Addiction has also been shown for other growth-regulating pathways, such as mTOR, WNT–TCF, and Notch, and in mouse models to c-MYC, and even miRNAs.
Oncogene addiction implies that despite the multistage nature of tumorigenesis, targeting of certain single oncogenes can be remarkably effective in cancer treatment. The shaking off of growth factor dependence is a critical step in tumorigenesis, but often carries with it the addiction to those signaling pathways that have enabled it. Cancers with activating mutations in key signaling pathways, particularly those in EGFR or related HER2, such as non-small-cell lung and breast cancers, become “addicted” to these signaling pathways. In other words, therapeutic targeting of these pathways inevitably has major effects on the cancer or at least until resistance supervenes. Thus HER2-amplified breast cancers are highly responsive to trastuzumab, and lung cancers with activating mutations in EGFR are highly responsive to TKIs or mAb targeting EGFR. Cancer cells undergo apoptosis, because such treatments inactivate key downstream signaling pathways, PI3K–AKT–mTOR and MEK–ERK, which support growth and survival. At least in lung cancer, PI3K inhibition downregulates Mcl-1 and MEK inhibition upregulates BIM, showing key roles for both downstream signaling pathways in response to TKI.
Cancer cells are not necessarily addicted to the same extent even within a single tumor; cellular heterogeneity as exemplified by cancer stem cells, dormant cancer cells, and even the surviving descendants of earlier clones that have been supplanted is matched by differences at a molecular level. These subpopulations will behave differently and respond differently to treatments as a result. Moreover, in some cases these cells may endure and, after a short-lived renunciation of replication, serve to revivify the cancer sometime after apparently successful treatments have been concluded.
At the risk of belaboring the point, imatinib has not only irrevocably altered the treatment of CML but also unimpeachably changed the whole course of drug development for cancer. However, the imatinib story also illustrates the shortcomings of targeted therapies. Firstly, not all cells respond fully, which is explained by the presence of an inherently resistant subpopulation of CML stem cells, unlike their progeny not addicted to BCR–ABL, which can repopulate the disease if imatinib is stopped. Thus, even in the presence of imatinib, CML stems and progenitors persist and may even continue expansion, at least in part through the supportive presence of a network of locally available cytokines. Secondly, with continued use of imatinib, cancer cells eventually redraft their molecular roadmap; the acquisition of resistance-conferring mutations allows cancer cells to escape from growth inhibition.
Non-Oncogene Addiction
Adversity has the effect of eliciting talents, which in prosperous circumstances would have lain dormant.
Horace
Maybe not surprisingly, cancer cells are also dependent on a number of key genes and proteins that are not oncogenes. These include regulators of translational control such as 4EBP–eIF4E, a variety of DNA repair genes such as PARP1, heat shock proteins (HSPs), metabolic enzymes, proteases such as Taspase1, and others needed alongside various oncogenic mutations. Scaffold proteins that may be crucial for supporting the structure and interactivity of growth-regulating pathways are likely also important in many aspects of cell signaling. Scaffold proteins are becoming of great interest in cancer biology as they contribute to a variety of key processes such as signaling kinetics, crosstalk between signaling pathways, and conversely buffering against this in some cases. Scaffolding also decreases reaction times by co-localizing enzymes and substrates. Thus, for example, the three successive levels of the RAF–MEK–ERK pathway operate within a scaffold provided by a kinase suppressor of RAS (KSR) proteins, and transformation by mutant RAS can be blocked by knocking out KSR1 as this prevents recruitment of BRAF.
This reliance of oncogenes on subordinate non-oncogenes is the basis of the “non-oncogene” addiction hypothesis, whereby a variety of key non-oncogenes is essential to tumor maintenance and therefore comprises attractive treatment targets. Thus, for example, breast cancers with defective homologous recombination (HR) or glioblastoma cells overexpressing an oncogenic variant of EGFR are hyperdependent on base excision repair (BER) genes such as PARP1. Some mTOR-mutated lymphomas require 4EBP–eIF4E cap-dependent translation and respond to drugs such as PP42 which target this protein.
HR is an important process in the repair of DNA double-strand breaks (DSBs). DSBs can arise through aberrant cancer cell cycles or indirectly from many of the DNA-damaging treatments such as radiotherapy that are directed against cancer. HR is largely restricted to dividing cells during the S and G2 phases of the cell cycle, so inhibiting this process will have comparatively less effect on normal cells. Moreover, as some cancers are bereft of alternative repair options, the resulting dependence on those remaining options generates the possibility for what is described as synthetic lethality. Combining the inhibition of two or more complementary pathways has a far greater effect than targeting either alone, as exemplified by PARP inhibition in BRCA-defective breast cancers (Chapter 10). This important area will be discussed in the “The concept of synthetic lethality” section.
The Concept of Synthetic Lethality and Collateral Vulnerability
Do we not wile away moments of inanity or fatigued waiting by repeating some trivial movement or sound, until the repetition has bred a want, which is incipient habit.
George Eliot
Sometimes it isn’t possible to target the actual mutant cancer-driving protein or to restore a missing anticancer protein. Mutant RAS, for example, has proved notoriously difficult to directly target with small-molecule drugs. An alternative is to exploit the fact that given mutations may render the cancer cell addicted to some other protein which might represent an easier target. A simple analogy would be that removing a parachute would have little effect on a person’s survival unless he or she had first been pushed out of an airplane.
Two genes are considered synthetic lethal if mutation of either alone does not compromise viability but mutation of both is fatal (Fig. 16.4). Synthetic lethality also occurs between genes and small molecules, and has been employed to study drug activity. In simple terms, targeting a gene that is synthetic lethal to a cancer-relevant mutation would be fatal only to cancer cells – another way of designing cancer-specific cytotoxic drugs. One scenario in which synthetic lethal interactions are likely to be critically important in cancer drug development is in the targeting of molecules that are synthetic lethal with loss of tumor suppressors.
Figure 16.4 Synthetic lethality. A targeted drug against the synthetic lethal has little effect on the cell with an intact alternative pathway but is lethal for the cell without it.
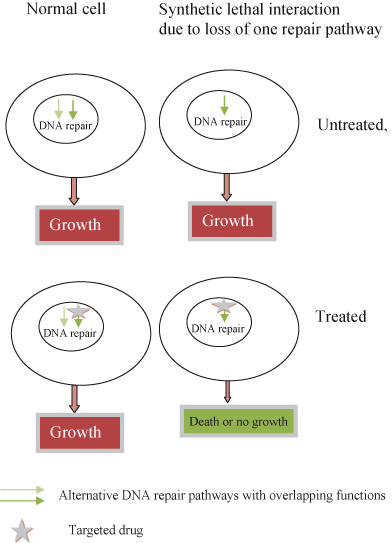
One recent example of this is BRCA1, which regulates HR-mediated DSB repair. Breast or ovarian cancers with mutated BRCA1 or BRCA2 survive this defect because they utilize another DNA repair pathway regulated by PARP1 to repair DSBs and allow replication fork progression. PARP inhibition kills cancer cells by allowing accumulation of DSBs and is conditionally “synthetic lethal” with the repair defect. Obviously, it is simpler to give a PARP inhibitor than to try to put BRCA back into every deficient cell.
Other synthetic lethal interactions are being screened across a range of model organisms, so this is unlikely to be the end of the story. Thus, loss of RAD52 is also synthetic lethal with BRCA2 and loss of Taspase-1 (threonine aspartase 1) with MYC and RAS in transformed cancer cells.
In addition to the DDR, cancer cells may depend on a range of “stress relievers” to prevent apoptosis or senescence when faced with potentially lethal oxidative stress, oncogenic stress, or altered metabolism. Drugs that incapacitate this “inner therapist” might be synthetic lethal for many cancer cells, with stress-inducing mutations such as deregulated MYC. Normal cells will not share this trauma, as exemplified by the synergism between chloroquine, an inhibitor of autophagy, and DNA damage in mice. Here, autophagy protects cancer cells from necrosis, but if autophagy is inhibited the cancer cell will die.
Another way to identify cancer cell vulnerabilities arises from the frequent presence of genomic instability. The premise is a simple one. The DDR for the loss of a known tumor suppressor will often also cause the “collateral” loss of other adjacent, or passenger, genes. If these other genes are critical and functionally redundant, their loss may render the cancer cell vulnerable in some previously unappreciated way. A number of such genes have been identified and given the acronym CYCLOPS (copy number alterations yielding cancer liabilities owing to partial loss) genes. This elite group include genes encoding spliceosome, proteasome, and ribosome components. One such gene, glycolytic gene enolase 1 (ENO1), is deleted in glioblastoma (GBM) but causes no obvious phenotype unless expression of ENO2 is also lost. This can be exploited therapeutically by administration of an enolase inhibitor, phosphonoacetohydroxamate, which is synthetic lethal in ENO1-deleted GBM cells.
Clinical Progress in Biological and Molecular Targeted Therapies
Emancipation from the bondage of the soil is no freedom for the tree.
Rabindranath Tagore
Given the importance of oncogene activation in human cancers, specific targeting of oncogenic pathways can be a highly effective therapeutic strategy. The approval in 2001 by the FDA of the drug imatinib (Gleevec) for the treatment of CML was a crucial milestone, because it was the first agent aimed at a specific cancer target (the BCR-ABL tyrosine kinase). Since then a myriad of new targeted agents, small molecule drugs and mAb, have reached the market, including such household names as trastuzumab.
Fig. 16.5 shows an overview of some of the most prominent targets of growth signaling pathways in new cancer drug development. Given, that many oncogenes are tyrosine kinases it is not surprising that most efforts have been directed against them.
BCR-ABL is a typical example of an aberrantly activated tyrosine kinase, discussed in more detail in Chapter 6. Briefly, a reciprocal translocation creates a transcript encoding a novel fusion protein known as BCR-ABL, a deregulated Abl (Abelson murine leukemia viral oncogene) kinase where the endogenous autoregulatory domain is replaced by sequences from BCR (breakpoint cluster region). Attempts to target the BCR-ABL protein resulted directly in the groundbreaking discovery of imatinib mesylate (see Box 16.1). Imatinib is now known to also inhibit other TKs including KIT, constitutively activated by c-KIT mutations in the rare and otherwise treatment-resistant gastrointestinal stromal tumurs (GISTs).
The success of imatinib has fuelled development of further kinase inhibitors as anticancer drugs (Table 16.2). As discussed in earlier chapters and below, cancer cells may become “addicted” to aberrant oncogenic signaling pathways and may as a result be especially sensitive to drugs inhibiting these pathways.
Table 16.2 Tyrosine kinase inhibitors
| Anti-ERBB family monoclonal antibodies (see also Chapter 5) | ||
| Drug | Target | Clinical status |
| Trastuzumab and herceptin | ERBB2 (HER2) ligand interactions and downregulation of receptor | |



