10
Bioinformatics & Computational Biology
Peter J. Kennelly, PhD & Victor W. Rodwell, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe the major features of genomics, proteomics, and bioinformatics.
Describe the major features of genomics, proteomics, and bioinformatics.
![]() Summarize the principal features and medical relevance of the Encode project.
Summarize the principal features and medical relevance of the Encode project.
![]() Describe the functions served by HapMap, Entrez Gene, BLAST, and the dbGAP databases.
Describe the functions served by HapMap, Entrez Gene, BLAST, and the dbGAP databases.
![]() Describe the major features of computer-aided drug design and discovery.
Describe the major features of computer-aided drug design and discovery.
![]() Describe possible future applications of computational models of individual pathways and pathway networks.
Describe possible future applications of computational models of individual pathways and pathway networks.
![]() Outline the possible medical utility of “virtual cells.”
Outline the possible medical utility of “virtual cells.”
BIOMEDICAL IMPORTANCE
The first scientific models of pathogenesis, such as Louis Pasteur’s seminal germ theory of disease, were binary in nature: each disease possessed a single, definable causal agent. Malaria was caused by the amoeba Plasmodium falciparum, tuberculosis by the bacterium Mycobacterium tuberculosis, sickle cell disease by a mutation in a gene encoding one of the subunits of hemoglobin, poliomyelitis by poliovirus, and scurvy by a deficiency in ascorbic acid. The strategy for treating or preventing disease thus could be reduced to a straightforward process of tracing the causal agent, and then devising some means of eliminating it, neutralizing its effects, or blocking its route of transmission. This approach has been successfully employed to understand and treat a wide range of infectious and genetic diseases. However, it has become clear that the determinants of many pathologies—including cancer, coronary heart disease, type II diabetes, and Alzheimer’s disease—are multifactorial in nature. Rather than having a specific causal agent or agents whose presence is both necessary and sufficient, the appearance and progression of the aforementioned diseases reflect the complex interplay between each individual’s genetic makeup, other inherited or epigenetic factors, and environmental factors such as diet, lifestyle, toxins, viruses, or bacteria.
The challenge posed by multifactorial diseases demands a quantum increase in the breadth and depth of our knowledge of living organisms capable of matching their sophistication and complexity. We must identify the many as yet unknown proteins encoded within the genomes of humans and the organisms with which they interact, their cellular functions and interactions. We must be able to trace the factors, both external and internal, that compromise human health and wellbeing by analyzing the impact of dietary, genetic, and environmental factors across entire communities or populations. The sheer mass of information that must be processed lies well beyond the ability of the human mind to review and analyze unaided. To understand, as completely and comprehensively as possible, the molecular mechanisms that underlie the behavior of living organisms, the manner in which perturbations can lead to disease or dysfunction, and how such perturbing factors spread throughout a population, biomedical scientists have turned to sophisticated computational tools to collect and evaluate biologic information on a mass scale.
GENOMICS: AN INFORMATION AVALANCHE
Physicians and scientists have long understood that the genome, the complete complement of genetic information of a living organism, represented a rich source of information concerning topics ranging from basic metabolism to evolution to aging. However, the massive size of the human genome, 3 × 109 nucleotide base pairs, required a paradigm shift in the manner in which scientists approached the determination of DNA sequences. Recent advances in bioinformatics and computational biology have, in turn, been fueled by the need to develop new approaches to “mine” the mass of sequence data generated by the application of increasingly efficient and economical technology to the genomes of hundreds of new organisms and, most recently, of several individual human beings.
The Human Genome Project
The successful completion of the Human Genome Project (HGP) represents the culmination of more than six decades of achievements in molecular biology, genetics, and biochemistry. The chronology below lists several of the milestone events that led to the determination of the entire sequence of the human genome.
1944—DNA is shown to be the hereditary material
1953—Concept of the double helix is posited
1966—The genetic code is solved
1972—Recombinant DNA technology is developed
1977—Practical DNA sequencing technology emerges
1983—The gene for Huntington’s disease is mapped
1985—The Polymerase Chain Reaction (PCR) is invented
1986—DNA sequencing becomes automated
1986—The gene for Duchenne muscular dystrophy is identified
1989—The gene for cystic fibrosis is identified
1990—The Human Genome Project is launched in the United States
1994—Human genetic mapping is completed 1996—The first human gene map is established 1999—The Single Nucleotide Polymorphism Initiative is started
1999—The first sequence of a human chromosome, number 22, is completed
2000—“First draft” of the human genome is completed 2003—Sequencing of the first human genome is completed
2010—Scientists embark on the sequencing of 1000 individual genomes to determine degree of genetic diversity in humans
By 2011 >180 eukaryotic, prokaryotic, and archaeal genomes had been sequenced. Examples include the genomes of chicken, cat, dog, elephant, rat, rabbit, orangutan, woolly mammoth, and duck-billed platypus, and the genomes of several individuals including Craig Venter and James Watson. The year 2010 saw completion of the Neanderthal genome, whose initial analysis suggests that up to 2% of the DNA in the genome of present-day humans outside of Africa originated in Neanderthals or in Neanderthal ancestors.
As of this writing, the genome sequences of >5,000 biological entities, ranging from viruses and bacteria to plants and animals have been reported. Ready access to genome sequences from organisms spanning all three phylogenetic domains and to the powerful algorithms requisite for manipulating and transforming the data derived from these sequences has transformed basic research in biology, microbiology, pharmacology, and biochemistry.
Genomes and Medicine
By comparing the genomes of pathogenic and nonpathogenic strains of a particular microorganism, genes likely to encode determinants of virulence can be highlighted by virtue of their presence in only the virulent strain. Similarly, comparison of the genomes of a pathogen with its host can identify genes unique to the former. Drugs targeting the protein products of the pathogen–specific genes should, in theory, produce little or no side effects for the infected host. The coming decade will witness the expansion of the “Genomics Revolution” into the day-to-day practice of medicine and agriculture as physicians and scientists exploit new knowledge of the human genome and of the genomes of the organisms that colonize, feed, and infect Homo sapiens. Whereas the first human genome project required several years, hundreds of people, and many millions of dollars to complete, quantum leaps in efficiency and economy have led one company to project that up to one million persons will have their individual genome sequences determined by the year 2014. The ability to diagnose and treat patients guided by knowledge of their genetic makeup, an approach popularly referred to as “designer medicine,” will render medicine safer and more effective.
Exome Sequencing
A harbinger of this new era has been provided by the application of “exome sequencing” to the diagnosis of rare or cryptic genetic diseases. The exome consists of those segments of DNA, called exons, that code for the amino acid sequences of proteins (Chapter 36). Since exons comprise only about 1% of the human genome, the exome represents a much smaller and more tractable target than the complete genome. Comparison of exome sequences has identified the genes responsible for a growing list of diseases that includes retinitis pigmentosa, Freeman–Sheldon syndrome, and Kabuki syndrome.
The Potential Challenges of Designer Medicine
While genome-based “designer medicine” promises to be very effective, it also confronts humanity with profound challenges in the areas of ethics, law, and public policy. Who owns and controls access to this information? Can a life or health insurance company, for instance, deny coverage to an individual based upon the risk factors apparent from their genome sequence? Does a prospective employer have the right to know a potential employee’s genetic makeup? Do prospective spouses have the right to know their fiancées’ genetic risk factors? Ironically, the resolution of these issues may prove a more lengthy and laborious process than did the determination of the first human genome sequence.
BIOINFORMATICS
Bioinformatics exploits the formidable information storage and processing capabilities of the computer to develop tools for the collection, collation, retrieval, and analysis of biologic data on a mass scale. That many bioinformatic resources (see below) can be accessed via the Internet provides them with global reach and impact. The central objective of a typical bio-informatics project is to assemble all of the available information relevant to a particular topic in a single location, often referred to as a library or database, in a uniform format that renders the data amenable to manipulation and analysis by computer algorithms.
Bioinformatic Databases
The size and capabilities of bioinformatic databases can vary widely depending upon the scope and nature of their objectives. The PubMed database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=pubmed) compiles citations for all articles published in thousands of journals devoted to biomedical and biological research. Currently, PubMed contains >20 million citations. By contrast, the RNA Helicase Database (http://www.rnahelicase.org/) confines itself to the sequence, structure, and biochemical and cellular function of a single family of enzymes, the RNA helicases.
Challenges of Database Construction
The construction of a comprehensive and user-friendly database presents many challenges. First, biomedical information comes in a wide variety of forms. For example, the coding information in a genome, although voluminous, is composed of simple linear sequences of four nucleotide bases. While the number of amino acid residues that define a protein’s primary structure is minute relative to the number of base pairs in a genome, a description of a protein’s x-ray structure requires that the location of each atom be specified in three-dimensional space.
Second, the designer must correctly anticipate the manner in which users may wish to search or analyze the information within a database, and devise algorithms for coping with these variables. Even the seemingly simple task of searching a gene database commonly employs, alone or in various combinations, criteria as diverse as the name of the gene, the name of the protein that it encodes, the biologic function of the gene product, a nucleotide sequence within the gene, a sequence of amino acids within the protein it encodes, the organism in which it is present, or the name of the investigators who determined the sequence of that gene.
EPIDEMIOLOGY ESTABLISHED THE MEDICAL POTENTIAL OF INFORMATION PROCESSING
The power of basic biomedical research resides in the laboratory scientist’s ability to manipulate homogenous, well-defined research targets under carefully controlled circumstances. The ability to independently vary the qualitative and quantitative characteristics of both target and input variables permits cause–effect relationships to be determined in a direct and reliable manner. These advantages are obtained, however, by employing “model” organisms such as mice or cultured human cell lines as stand-ins for the human patients that represent the ultimate targets for, and beneficiaries of, this research. Laboratory animals do not always react as do Homo sapiens, nor can a dish of cultured fibroblast, kidney, or other cells replicate the incredible complexity of a human being.
Although unable to conduct rigorously controlled experiments on human subjects, careful observation of real world behavior has long proven to be a source of important biomedical insights. Hippocrates, for example, noted that while certain epidemic diseases appeared in a sporadic fashion, endemic diseases such as malaria exhibited clear association with particular locations, age group, etc. Epidemiology refers to the branch of the biomedical sciences that employs bioinformatic approaches to extend our ability and increase the accuracy with which we can identify factors that contribute to or detract from human health through the study of real world populations.
Early Epidemiology of Cholera
One of the first recorded epidemiological studies, conducted by Dr. John Snow, employed simple geospatial analysis to track the source of a cholera outbreak. Epidemics of cholera, typhus, and other infectious diseases were relatively common in the crowded, unsanitary conditions of nineteenth century London. By mapping the locations of the victims’ residences, Snow was able to trace the source of the contagion to the contamination of one of the public pumps that supplied citizens with their drinking water (Figure 10–1). Unfortunately, the limited capacity of hand calculations or graphing rendered the success of analyses such as Snow’s critically dependent upon the choice of the working hypothesis used to select the variables to be measured and processed. Thus, while nineteenth century Londoners also widely recognized that haberdashers were particularly prone to display erratic and irrational behavior (eg, “as Mad as a Hatter”), nearly a century would pass before the cause was traced to the mercury compounds used to prepare the felt from which the hats were constructed.
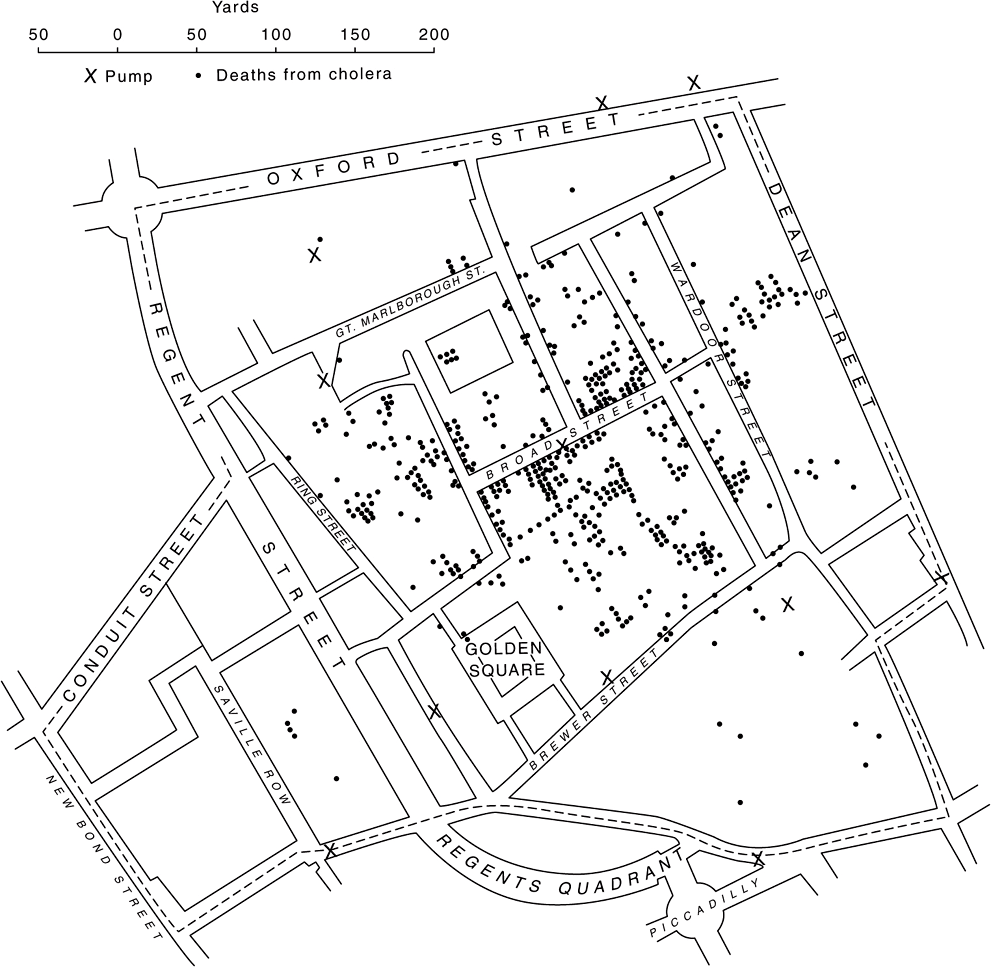
FIGURE 10–1 This version of the map drawn by Dr. John Snow compares the location of the residences of victims of an 1854 London cholera epidemic (Dots) with the locations of the pumps that supplied their drinking water (X’s). Contaminated water from the pump on Broad Street, lying roughly in the center of the cluster of victims, proved to be the source of the epidemic in this neighborhood.
Impact of Bioinformatics on Epidemiological Analysis
As the process of data analysis has become automated, the sophistication and success rate of epidemiological analyses has risen accordingly. Today, complex computer algorithms enable researchers to assess the influence of a broad range of health-related parameters when tracking the identity and source or reconstructing the transmission of a disease or condition: height; weight; age; gender; body mass index; diet; ethnicity; medical history; profession; drug, alcohol, or tobacco use; exercise; blood pressure; habitat; marital status; blood type; serum cholesterol level; etc. Equally important, modern bioinformatics may soon enable epidemiologists to dissect the identities and interactions of the multiple factors underlying complex diseases such as cancer or Alzheimer’s disease.
The accumulation of individual genome sequences will shortly introduce a powerful new dimension to the host of biological, environmental, and behavioral factors to be compared and contrasted with each person’s medical history. One of the first fruits of these studies has been the identification of genes responsible for a few of the >3000 known or suspected Mendelian disorders whose causal genetic abnormalities have yet to be traced. The ability to evaluate contributions of and the interactions among an individual’s genetic makeup, behavior, environment, diet, and lifestyle holds the promise of eventually revealing the answers to the age-old question of why some persons exhibit greater vitality, stamina, longevity, and resistance to disease than others—in other words, the root sources of health and wellness.
BIOINFORMATIC AND GENOMIC RESOURCES
The large collection of databases that have been developed for the assembly, annotation, analysis and distribution of biological and biomedical data reflects the breadth and variety of contemporary molecular, biochemical, epidemiological, and clinical research. Below are listed examples of the following prominent bioinformatics resources: UniProt, GenBank, and the Protein Database (PDB) represent three of the oldest and most widely used bioinformatics databases. Each complements the other by focusing on a different aspect of macromolecular structure.
Uniprot
The UniProt Knowledgebase (http://www.pir.uniprot.org/) can trace its origins to the Atlas of Protein Sequence and Structure, a printed encyclopedia of protein sequences first published in 1968 by Margaret Dayhoff and the National Biomedical Research Foundation at Georgetown University. The aim of the Atlas was to facilitate studies of protein evolution using the amino acid sequences being generated consequent to the development of the Edman method for protein sequencing (Chapter 4). In partnership with the Martinsreid Institute for Protein Sequences and the International Protein Information Database of Japan, the Atlas made the transition to electronic form as the Protein Information Resource (PIR) Protein Sequence Database in 1984. In 2002, PIR integrated its database of protein sequence and function with the Swiss-Prot Protein Database established by Amos Bairoch in 1986 under the auspices of the Swiss Institute of Bioinformatics and the European Bioinformatics Institute, to form the world’s most comprehensive resource on protein structure and function, the UniProt Knowledgebase.
GenBank
The goal of GenBank (http://www.ncbi.nlm.nih.gov/Genbank/), the National Institutes of Health’s (NIH) genetic sequence database, is to collect and store all known biological nucleotide sequences and their translations in a searchable form. Established in 1979 by Walter Goad of Los Alamos National Laboratory, GenBank currently is maintained by the National Center for Biotechnology Information at the NIH. GenBank constitutes one of the cornerstones of the International Sequence Database Collaboration, a consortium that includes the DNA Database of Japan and the European Molecular Biology Laboratory.
PDB
The RCSB Protein Data Base (PDB) (http://www.rcsb.org/pdb/home/home.do), a repository of the three-dimensional structures of proteins, polynucleotides, and other biological macromolecules, was established in 1971 by Edgar Meyer and Walter Hamilton of Brookhaven National Laboratories. In 1998, responsibility for the PDB was transferred to the Research Collaboration for Structural Bioinformatics formed by Rutgers University, the University of California at San Diego, and the University of Wisconsin. The PDB contains well over 50,000 three-dimensional structures for proteins, as well as proteins bound with substrates, substrate analogs, inhibitors, or other proteins. The user can rotate these structures freely in three-dimensional space, highlight specific amino acids, and select from a variety of formats such as space filling, ribbon, backbone, etc. (Chapters 5, 6, and 10).
SNPs & Tagged SNPs
While the genome sequence of any two individuals is 99.9% identical, human DNA contains ~10 million sites where individuals differ by a single-nucleotide base. These sites are called Single-Nucleotide Polymorphisms or SNPs. When sets of SNPs localized to the same chromosome are inherited together in blocks, the pattern of SNPs in each block is termed a hap-lotype. By comparing the haplotype distributions between groups of individuals that differ in some physiological characteristic, such as susceptibility to a disease, biomedical scientists can identify SNPs that are associated with specific phenotypic traits. This process can be facilitated by focusing on Tag SNPs, a subset of the SNPs in a given block sufficient to provide a unique marker for a given haplotype. Detailed study of each region should reveal variants in genes that contribute to a specific disease or response.
HapMap
In 2002, scientists from the United States, Canada, China, Japan, Nigeria, and the United Kingdom launched the International Haplotype Map (HapMap) Project (http://hapmap.ncbi.nlm.nih.gov/), a comprehensive effort to identify SNPs associated with common human diseases and differential responses to pharmaceuticals. The resulting HapMap Database should lead to earlier and more accurate diagnosis, improved prevention, and patient management. Knowledge of an individual’s genetic profile will also be used to guide the selection of safer and more effective drugs or vaccines, a process termed pharmacogenomics. These genetic markers will also provide labels with which to identify and track specific genes as scientists seek to learn more about the critical processes of genetic inheritance and selection.
ENCODE
Identification of all the functional elements of the genome will vastly expand our understanding of the molecular events that underlie human development, health, and disease. To address this goal, in late 2003, the National Human Genome Research Institute (NHGRI) initiated the ENCODE (Encyclopedia of DNA Elements) Project. Based at the University of California at Santa Cruz, ENCODE (http://www.genome.gov/10005107) is a collaborative effort that combines laboratory and computational approaches to identify every functional element in the human genome. Consortium investigators with diverse backgrounds and expertise collaborate in the development and evaluation of new high-throughput techniques, technologies, and strategies to address current deficiencies in our ability to identify functional elements.
The pilot phase of ENCODE targeted ~1% (30 Mb) of the human genome for rigorous computational and experimental analysis. A variety of methods were employed to identify, or annotate, the function of each portion of the DNA in 500 base pair steps. These pilot studies revealed that the human genome contains a large number and variety of functionally active components interwoven to form complex networks. The successful prosecution of this pilot study has resulted in the funding of a series of Scale-Up Projects aimed at tackling the remaining 99% of the genome.
Entrez Gene
Entrez Gene (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene), a database maintained by the National Center for Biotechnology Information (NCBI), provides a variety of information about individual human genes. The information includes the sequence of the genome in and around the gene, exon-intron boundaries, the sequence of the mRNA(s) produced from the gene, and any known phenotypes associated with a given mutation of the gene in question. Entrez Gene also lists, where known, the function of the encoded protein and the impact of known single-nucleotide polymorphisms within its coding region.
dbGAP
dbGAP, the Database of Genotype and Phenotype, is an NCBI database that complements Entrez Gene (http://www.ncbi.nlm.nih.gov/gap). dbGAP compiles the results of research into the links between specific genotypes and phenotypes. To protect the confidentiality of sensitive clinical data, the information contained in dbGAP is organized into open- and controlled-access sections. Access to sensitive data requires that the user apply for authorization to a data access committee.
Additional Databases
Other databases dealing with human genetics and health include OMIM, Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim), HGMD, the HumanGene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), the Cancer Genome Atlas (http://cancergenome.nih.gov/), and GeneCards (http://www.genecards.org/), which tries to collect all relevant information on a given gene from databases worldwide to create a single, comprehensive “card” for each.
COMPUTATIONAL BIOLOGY
The primary objective of computational biology is to develop computer models that apply physical, chemical, and biological principles to reproduce the behavior of biologic molecules and processes. Unlike bioinformatics, whose major focus is the collection and evaluation of existing data, computational biology is experimental and exploratory in nature. By performing virtual experiments and analyses “in silico,” meaning performed on a computer or through a computer simulation, computational biology offers the promise of greatly accelerating the pace and efficiency of scientific discovery.
Computational biologists are attempting to develop predictive models that will (1) permit the three-dimensional structure of a protein to be determined directly from its primary sequence, (2) determine the function of unknown proteins from their sequence and structure, (3) screen for potential inhibitors of a protein in silico, and (4) construct virtual cells that reproduce the behavior and predict the responses of their living counterparts to pathogens, toxins, diet, and drugs. The creation of computer algorithms that accurately imitate the behavior of proteins, enzymes, cells, etc., promises to enhance the speed, efficiency, and the safety of biomedical research. Computational biology will also enable scientists to perform experiments in silico whose scope, hazard, or nature renders them inaccessible to, or inappropriate for, conventional laboratory or clinical approaches.
IDENTIFICATION OF PROTEINS BY HOMOLOGY
One important method for the identification, also called annotation, of novel proteins and gene products compares the sequences of novel proteins with those of proteins whose functions or structures had been determined. Simply put, homology searches and multiple sequence comparisons operate on the principle that proteins that perform similar functions will share conserved domains or other sequence features or motifs, and vice versa (Figure 10–2). Of the many algorithms developed for this purpose, the most widely used is BLAST.

FIGURE 10–2 Representation of a multiple sequence alignment. Languages evolve in a fashion that mimics that of genes and proteins. Shown is the English word “physiological” in several languages. The alignment demonstrates their conserved features. Identities with the English word are shown in dark red; linguistic similarities in dark blue. Multiple sequence alignment algorithms identify conserved nucleotide and amino acid letters in DNA, RNA, and polypeptides in an analogous fashion.
BLAST
BLAST (Basic Local Alignment Search Tool) and other sequence comparison/alignment algorithms trace their origins to the efforts of early molecular biologists to determine whether observed similarities in sequence among proteins that perform parallel metabolic functions were indicative of progressive changes in a common ancestral protein. The major evolutionary question addressed was whether the similarities reflected (1) descent from a common ancestral protein (divergent evolution) or (2) the independent selection of a common mechanism for meeting some specific cellular need (convergent evolution), as would be anticipated if one particular solution was overwhelmingly superior to the alternatives. Calculation of the minimum number of nucleotide changes required to interconvert putative protein isoforms allows inferences to be drawn concerning whether or not the similarities and differences exhibit a pattern indicative of progressive change from a shared origin.
BLAST has evolved into a family of programs optimized to address specific needs and data sets. Thus, blastp compares an amino acid query sequence against a protein sequence database, blastn compares a nucleotide query sequence against a nucleotide sequence database, blastx compares a nucleotide query sequence translated in all reading frames against a protein sequence database to reveal potential translation products, tblastn compares a protein query sequence against a nucleotide sequence database dynamically translated in all six reading frames, and tblastx compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database. Unlike multiple sequence alignment programs that rely on global alignments, the BLAST algorithms emphasize regions of local alignment to detect relationships among sequences with only isolated regions of similarity. This approach provides speed and increased sensitivity for distant sequence relationships. Input or “query” sequences are broken into “words” (default size 11 for nucleotides, 3 for amino acids). Word hits to databases are then extended in both directions.
IDENTIFICATION OF “UNKNOWN” PROTEINS
A substantial portion of the genes discovered by genome sequencing projects code for “unknown” or hypothetical polypeptides for which homologs of known function are lacking. Bioinformaticists are developing tools to enable scientists to deduce the three-dimensional structure and function of cryptic proteins directly from their amino acid sequences. Currently, the list of unknown proteins uncovered by genomics contains thousands of entries, with new entries being added as more genome sequences are solved. The ability to generate structures and infer function in silico promises to significantly accelerate protein identification and provide insight into the mechanism by which proteins fold. This knowledge will aid in understanding the underlying mechanisms of various protein folding diseases, and will assist molecular engineers to design new proteins to perform novel functions.
The Folding Code
Comparison of proteins whose three-dimensional structures have been determined by x-ray crystallography or NMR spectroscopy can reveal patterns that link specific primary sequence features to specific primary, secondary, and tertiary structures—sometimes called the folding code. The first algorithms used the frequency with which individual amino acids occurred in α-helices, β-sheets, turns, and loops to predict the number and location of these elements within the sequence of a polypeptide, known as secondary structure topography. By extending this process, for example, by weighing the impact of hydrophobic interactions in the formation of the protein core, algorithms of remarkable predictive reliability are being developed. However, while current programs perform well in generating the conformations of proteins comprised of a single domain, projecting the likely structure of membrane proteins and those composed of multiple domains remains problematic.
Relating Three-Dimensional Structure to Function
Scientists are also attempting to discern patterns of three-dimensional structure that correlate to specific physiologic functions. The space-filling representation of the enzyme HMG-CoA reductase and its complex with the drug lovastatin (Figure 10–3) provides some perspective on the challenges inherent in identifying ligand-binding sites from scratch. Where a complete three-dimensional structure can be determined or predicted, the protein’s surface can be scanned for the types of pockets and crevices indicative of likely binding sites for substrates, allosteric effectors, etc., by any one of a variety of methods such as tracing its surface with balls of a particular dimension (Figure 10–4). Surface maps generated with the program Graphical Representation and Analysis of Surface Properties, commonly referred to as GRASP diagrams, highlight the locations of neutral, negatively charged, and positively charged functional groups on a protein’s surface (Figure 10–5) to infer a more detailed picture of the biomolecule that binds to or “docks” at that site. The predicted structure of the ligands that bind to an unknown protein, along with other structural characteristics and sequence motifs can then provide scientists with the clues needed to make an “educated guess” regarding its biological function(s).
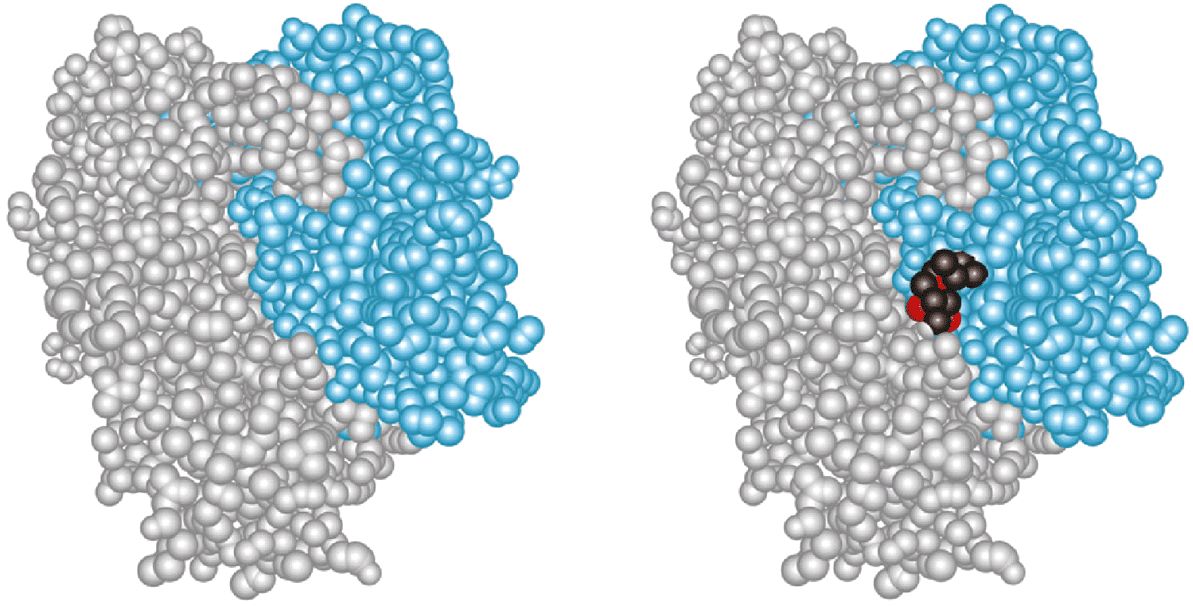
FIGURE 10–3 Shown are space-filling representations of the homodimeric HMG-CoA reductase from Pseudomonas mevalonii with (right) and without (left) the statin drug lovastatin bound. Each atom is represented by a sphere the size of its van der Waals’ radius. The two polypeptide chains are colored gray and blue. The carbon atoms of lovastatin are colored black and the oxygen atoms red. Compare this model with the backbone representations of proteins shown in Chapters 5 and 6. (Adapted from Protein Data Bank ID no. 1t02.)
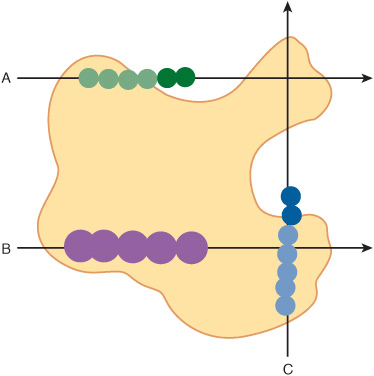
FIGURE 10–4 A simplified representation of a ligand site prediction program. Ligand site prediction programs such as POCKET, LIGSITE, or Pocket-Finder convert the three-dimensional structure of a protein into a set of coordinates for its component atoms. A two-dimensional slice of the space filled by these coordinates is presented as an irregularly shaped outline (yellow). A round probe is then passed repeatedly through these coordinates along a series of lines paralleling each of the three coordinate axes (A, B, C). Lightly shaded circles represent positions of the probe where its radius overlaps with one or more atoms in the Cartesian coordinate set. Darkly shaded circles represent positions where no protein atom coordinates fall within the probe’s radius. In order to qualify as a pocket or crevice within the protein, and not just open space outside of it, the probe must eventually encounter protein atoms lying on the other side of the opening (C).
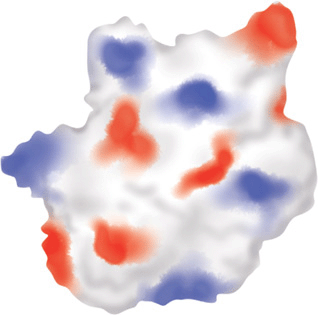
FIGURE 10–5 Representation of a GRASP diagram indicating the electrostatic topography of a protein. Shown is a space-filling representation of a hypothetical protein. Areas shaded in red indicate the presence of amino acid side chains or other moieties on the protein surface predicted to bear a negative charge at neutral pH. Blue indicates the presence of predicted positively charged groups. White denotes areas predicted to be electrostatically neutral.
COMPUTER-AIDED DRUG DESIGN
Computer-Aided Drug Design (CADD) employs the same type of molecular-docking algorithms used to identify ligands for unknown proteins. However, in this case the set of potential ligands to be considered is not confined to those occurring in nature and is aided by empirical knowledge of the structure or functional characteristics of the target protein.
Molecular Docking Algorithms
For proteins of known three-dimensional structure, molecular-docking approaches employ programs that attempt to fit a series of potential ligand “pegs” into a designated binding site or “hole” on a protein template. To identify optimum ligands, docking programs must account for matching shapes as well as the presence and position of complementary hydrophobic, hydrophilic, and charge attributes. The binding affinities of the inhibitors selected on the basis of early docking studies were disappointing, as the rigid models for proteins and ligands employed were incapable of replicating the conformational changes that occur in both ligand and protein as a consequence of binding, a phenomenon referred to as “induced fit” (Chapter 7).
Imbuing proteins and ligands with conformational flexibility requires massive computing power, however. Hybrid approaches have thus evolved that employ a set, or ensemble, of templates representing slightly different conformations of the protein (Figure 10–6) and either ensembles of ligand conformers (Figure 10–7) or ligands in which only a few select bonds are permitted to rotate freely. Once the set of potential ligands has been narrowed, more sophisticated docking analyses can be undertaken to identify high-affinity ligands able to interact with their protein target across the latter’s spectrum of conformational states.
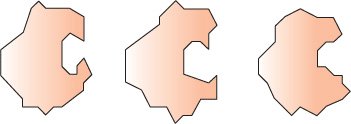
FIGURE 10–6 Two-dimensional representation of a set of conformers of a protein. Notice how the shape of the binding site changes.
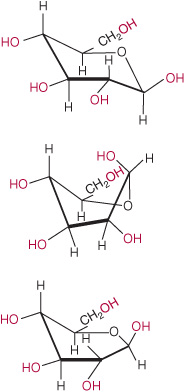
FIGURE 10–7 Conformers of a simple ligand. Shown are three of the many different conformations of glucose, commonly referred to as chair (Top), twist boat (Middle), and half chair (Bottom). Note the differences not only in shape and compactness but in the position of the hydroxyl groups, potential participants in hydrogen bonds, as highlighted in red.
Structure–Activity Relationships
If no structural template is available for the protein of interest, computers can be used to assist the search for high-affinity inhibitors by calculating and projecting Structure–Activity Relationships (SARs). In this process, the measured binding affinities for several known inhibitors are compared and contrasted to determine whether specific chemical features make positive or negative thermodynamic contributions to ligand binding. This information can then be used to search databases of chemical compounds to identify those which possess the most promising combination of positive versus negative features.
SYSTEMS BIOLOGY & VIRTUAL CELLS
The Goal of Systems Biology Is to Construct Molecular Circuit Diagrams
What if a scientist could detect, in a few moments, the effect of inhibiting a particular enzyme, of replacing a particular gene, the response of a muscle cell to insulin, the proliferation of a cancer cell, or the production of beta amyloid by entering the appropriate query into a computer? The goal of systems biology is to construct the molecular equivalent of circuit diagrams that faithfully depict the components of a particular functional unit and the interactions between them in logical or mathematical terms. These functional units can range in size and complexity from the enzymes and metabolites within a biosynthetic pathway to the network of proteins that controls the cell division cycle to, ultimately, entire cells, organs, and organisms. These models can then be used to perform “virtual” experiments that can enhance the speed and efficiency of empirical investigations by identifying the most promising lines of investigation and assisting in the evaluation of results. Perhaps more significantly, the ability to conduct virtual experiments extends the reach of the investigator, within the limits of the accuracy of the model, beyond the reach of current empirical technology.
Already, significant progress is being made. By constructing virtual molecular networks, scientists have been able to determine how cyanobacteria assemble a reliable circadian clock using only four proteins. Models of the T cell receptor signaling pathway have revealed how its molecular circuitry has been arranged to produce switch-like responses upon stimulation by agonist peptide-major histocompatability complexes (MHC) on an antigen-presenting cell. Scientists can use the gaps encountered in modeling molecular and cellular systems to guide the identification and annotation of the remaining protein pieces, in the same way that someone who solves a jigsaw puzzle surveys the remaining pieces for matches to the gaps in the puzzle. This reverse engineering approach has been successfully used to define the function of type II glycerate 2-kinases in bacteria and to identify “cryptic” folate synthesis and transport genes in plants.
Virtual Cells
Recently, scientists have been able to successfully create a sustainable metabolic network, composed of nearly two hundred proteins—an important step toward the creation of a virtual cell. The “holy grail” of systems biologists is to replicate the behavior of living human cells in silico. The potential benefits of such virtual cells are enormous. Not only will they permit optimum sites for therapeutic intervention to be identified in a rapid and unbiased manner, but unintended side effects may be revealed prior to the decision to invest time and resources in the synthesis, analysis, and trials of a potential pharmacophore. The ability to conduct fast, economic toxicological screening of materials ranging from herbicides to cosmetics will benefit human health. Virtual cells can also aid in diagnosis. By manipulating a virtual cell to reproduce the metabolic profile of a patient, underlying genetic abnormalities may be revealed. The interplay of the various environmental, dietary, and genetic factors that contribute to multifactorial diseases such as cancer can be systematically analyzed. Preliminary trials of potential gene therapies can be assessed safely and rapidly in silico.
The duplication of a living cell in silico represents an extremely formidable undertaking. Not only must the virtual cell possess all of the proteins and metabolites for the type of cell to be modeled (eg, from brain, liver, nerve, muscle, or adipose), but these must be present in the appropriate concentration and subcellular location. The model must also account for the functional dynamics of its components, binding affinities, catalytic efficiency, covalent modifications, etc. To render a virtual cell capable of dividing or differentiating will entail a further quantum leap in complexity and sophistication.
Molecular Interaction Maps Employ Symbolic Logic
The models constructed by systems biologists can take a variety of forms depending upon the uses for which they are intended and the data available to guide their construction. If one wishes to model the flux of metabolites through an anabolic or catabolic pathway, it is not enough to know the identities and the reactants involved in each enzyme-catalyzed reaction. To obtain mathematically precise values, it is necessary to know the concentrations of the metabolites in question, the quantity of each of each enzyme present, and their catalytic parameters.
For most users, it is sufficient that a model describe and predict the qualitative nature of the interactions between components. Does an allosteric ligand activate or inhibit the enzyme? Does dissociation of a protein complex lead to the degradation of one or more of its components? For this purpose, a set of symbols depicting the symbolic logic of these interactions was needed. Early representations frequently used the symbols previously developed for constructing flow charts (computer programming) or electronic circuits (Figure 10–8, top). Ultimately, however, systems biologists designed dedicated symbols (Figure 10–8, bottom) to depict these molecular circuit diagrams, more commonly referred to as Molecular Interaction Maps (MIM), an example of which is shown in Figure 10–9. Unfortunately, as is the case with enzyme nomenclature (Chapter 7) a consistent, universal set of symbols has yet to emerge.
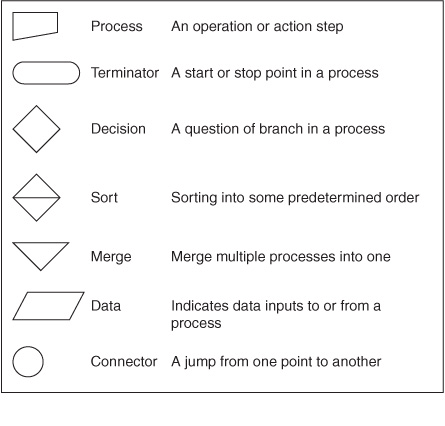
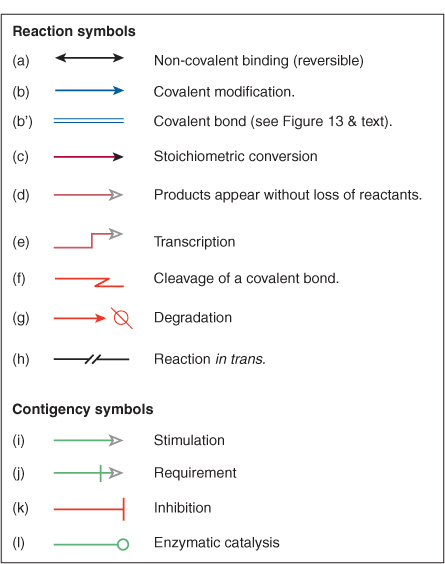
FIGURE 10–8 Symbols used to construct molecular circuit diagrams in systems biology. (Top) Sample flowchart symbols. (Bottom) Graphical symbols for molecular interaction maps (Adapted from Kohn KW et al: Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol Biol Cell 2006;17:1).
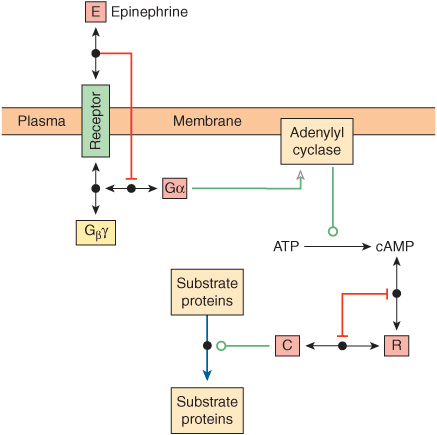
FIGURE 10–9 Representation of a molecular interaction network (MIN) depicting a signal transduction cascade leading to the phosphorylation of substrate proteins by the catalytic subunit, C, of the cyclic AMP-dependent protein kinase in response to epinephrine. Proteins are depicted as rectangles or squares. Double headed arrows indicate formation of noncovalent complex represented by dot at the midpoint of the arrow. Red lines with T-shaped heads indicate inhibitory interaction. Green arrow with hollow head indicates a stimulatory interaction. Green line with open circle at end indicates catalysis. Blue arrow with P indicates covalent modification by phosphorylation. (Symbols adapted from Kohn KW et al: Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol Biol Cell 2006;17:1.)
CONCLUSION
The rapidly evolving fields of bioinformatics and computational biology hold unparalleled promise for the future of both medicine and basic biology. Some promises are at present perceived clearly, others dimly, while yet others remain unimagined. A major objective of computational biologists is to develop computational tools that will enhance the efficiency, effectiveness, and speed of drug development. Epidemiologists employ computers to extract patterns within a population indicative of specific causes of and contributors to both disease and wellness. There seems little doubt that their impact on medical practice in the 21st century will equal or surpass that of the discovery of bacterial pathogenesis in the 19th century.
SUMMARY
![]() Genomics has yielded a massive quantity of information of great potential value to scientists and physicians.
Genomics has yielded a massive quantity of information of great potential value to scientists and physicians.
![]() Bioinformatics involves the design of computer algorithms and construction of databases that enable biomedical scientists to access and analyze the growing avalanche of biomedical data.
Bioinformatics involves the design of computer algorithms and construction of databases that enable biomedical scientists to access and analyze the growing avalanche of biomedical data.
![]() The objective of epidemiology is to extract medical insights from the behavior of heterogeneous human populations by the application of sophisticated statistical tools.
The objective of epidemiology is to extract medical insights from the behavior of heterogeneous human populations by the application of sophisticated statistical tools.
![]() Major challenges in the construction of user-friendly databases include devising means for storing and organizing complex data that accommodate a wide range of potential search criteria.
Major challenges in the construction of user-friendly databases include devising means for storing and organizing complex data that accommodate a wide range of potential search criteria.
![]() The goal of the Encode Project is to identify all the functional elements within the human genome.
The goal of the Encode Project is to identify all the functional elements within the human genome.
![]() The HapMap, Entrez Gene, and dbGAP databases contain data concerning the relation of genetic mutations to pathological conditions.
The HapMap, Entrez Gene, and dbGAP databases contain data concerning the relation of genetic mutations to pathological conditions.
![]() Computational biology uses computer algorithms to identify unknown proteins and conduct virtual experiments.
Computational biology uses computer algorithms to identify unknown proteins and conduct virtual experiments.
![]() BLAST is used to identify unknown proteins and genes by searching for sequence homologs of known function.
BLAST is used to identify unknown proteins and genes by searching for sequence homologs of known function.
![]() Computational biologists are developing programs that will predict the three-dimensional structure of proteins directly from their primary sequence.
Computational biologists are developing programs that will predict the three-dimensional structure of proteins directly from their primary sequence.
![]() Computer-aided drug design speeds drug discovery by trying to dock potential inhibitors to selected protein targets in silico.
Computer-aided drug design speeds drug discovery by trying to dock potential inhibitors to selected protein targets in silico.
![]() A major goal of systems biologists is to create faithful models of individual pathways and networks in order to elucidate functional principles and perform virtual experiments.
A major goal of systems biologists is to create faithful models of individual pathways and networks in order to elucidate functional principles and perform virtual experiments.
![]() The ultimate goal of systems biologists is to create virtual cells that can be used to more safely and efficiently diagnose and treat diseases, particularly those of a multifactorial nature.
The ultimate goal of systems biologists is to create virtual cells that can be used to more safely and efficiently diagnose and treat diseases, particularly those of a multifactorial nature.
![]() Systems biologists commonly construct schematic representations known as molecular interaction maps in which symbolic logic is employed to illustrate the relationships between the components making up a pathway or some other functional unit
Systems biologists commonly construct schematic representations known as molecular interaction maps in which symbolic logic is employed to illustrate the relationships between the components making up a pathway or some other functional unit
REFERENCES
Altschul SF, Gish W, Miller W, et al: Basic local alignment search tool. J Mol Biol 1990;215:403.
Collins FS, Barker AD: Mapping the human cancer genome. Sci Am 2007;296:50.
Collins FS, Green ED, Guttmacher AE, et al: A vision for the future of genomics research. A blueprint for the genomic era. Nature 2003;422:835.
Couzin J: The HapMap gold rush: researchers mine a rich deposit. Science 2006;312:1131.
Cravatt BF, Wright AT, Kozarich JW: Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem 2008;77:383.
Debes JD, Urrutia R: Bioinformatics tools to understand human diseases. Surgery 2004;135:579.
Dunning Hotopp JC, Grifantini R, Kumar N, et al: Comparative genomics of Neisseria meningitides: core genome, islands of horizontal transfer and pathogen-specific genes. Microbiology 2006;152:3691.
Ekins S, Mestres J, Testa B: In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol 2007;152:21.
Ekins S, Mestres J, Testa B: In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol 2007;152:9.
Kaiser J: Affordable ‘exomes’ fill gaps in a catalog of rare diseases. Science 2010;330:903.
Kim JH: Bioinformatics and genomic medicine. Genet Med 2002;4:62S.
Kohn KW, Aladjem MI, Weinstein JN, et al: Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol Biol Cell 2006;17:1.
Koonin EV, Galperin MY: Sequence—Evolution—Function. Computational Approaches to Comparative Genomics. Kluwer Academic Publishers, 2003.
Laurie ATR, Jackson RM: Methods for prediction of protein–ligand binding sites for structure-based drug design and virtual ligand screening. Curr Prot Peptide Sci 2006;7:395–406.
McInnes C: Virtual screening strategies in drug discovery. Curr Opin Cell Biol 2007;11:494.
Nebert DW, Zhang G, Vesell ES: From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab Rev 2008;40:187.
Sansom C: Genes and disease. The Scientist 2008;30:34. Slepchenko BM, Schaff JC, Macara I, et al: Quantitative cell biology with the Virtual Cell. Trends Cell Biol 2003;13:570.
Sudmant PH, Kitzman JO, Antonacci F, et al: Diversity of human gene copy number variation and multicopy genes. Science 2010;330:641.
Villoutreix BO, Renault N, Lagorce D, et al: Free resources to assist structure-based virtual ligand screening experiments. Curr Protein Pept Sci 2007;8:381.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


