57
Biochemical Case Histories
Robert K. Murray, MD, PhD & Peter L. Gross, MD, MSc, FRCP(C)
OBJECTIVES
After studying this chapter, you should be able to:
![]() Appreciate the importance of a sound knowledge of Biochemistry and Genetics in understanding and managing many clinical conditions.
Appreciate the importance of a sound knowledge of Biochemistry and Genetics in understanding and managing many clinical conditions.
![]() Understand the general features and some aspects of management of the following conditions: adenosine deaminase deficiency; Alzheimer disease; cholera; colorectal cancer; cystic fibrosis; diabetic ketoacidosis; Duchenne muscular dystrophy; acute ethanol intoxication; acute gout; hereditary hemochromatosis; primary hypothyroidism; kwashiorkor and protein-energy malnutrition; myocardial infarction; obesity; primary osteoporosis; xeroderma pigmentosum.
Understand the general features and some aspects of management of the following conditions: adenosine deaminase deficiency; Alzheimer disease; cholera; colorectal cancer; cystic fibrosis; diabetic ketoacidosis; Duchenne muscular dystrophy; acute ethanol intoxication; acute gout; hereditary hemochromatosis; primary hypothyroidism; kwashiorkor and protein-energy malnutrition; myocardial infarction; obesity; primary osteoporosis; xeroderma pigmentosum.
INTRODUCTION
In this final chapter, 16 case histories are presented and discussed. They illustrate the importance of knowledge of Biochemistry for the understanding of disease. Of course, as has been shown throughout the text, Biochemistry is also crucial for the understanding of health and wellness.
Most of the diseases discussed here are prevalent, or relatively prevalent, in a global sense. (Prevalence is the proportion of persons in a given population that has a particular disease at a point or interval of time.) However, two (xeroderma pigmentosum and severe combined immunodeficiency disease due to deficiency of adenosine deaminase [ADA]) are relatively rare. They are included because they illustrate two crucial biologic facts: the importance of DNA repair and of the immune system as protective mechanisms. In addition, ADA deficiency is the first disease for which gene therapy was performed in humans.
The reference values for laboratory tests cited in the cases below may differ from these listed by laboratories with which the reader may be familiar. This is because reference values from different laboratories vary somewhat, in part due to different methodologies. In this chapter, laboratory results are generally given as SI units (Systeme International d’Unites). Chapter 56 presents some of the basic principles concerned with the ordering and interpretation of laboratory tests. Table 56-11 lists the majority of the normal values for the lab tests referred to in this chapter, and gives both SI and “conventional” values (as widely used in the United States).
The doses of drugs administered in the treatment of the cases described here are not generally given; the reader should check these out on her/his own.
CASE 1: ADENOSINE DEAMINASE (ADA) DEFICIENCY CAUSING SEVERE COMBINED IMMUNODEFICIENCY DISEASE (SCID)
Causation
Genetic (due to mutations in the gene encoding ADA). Deficiency of ADA affects the immune system.
History and Physical Examination
A young girl aged 11 months was brought by her parents to a children’s hospital. She had had a number of attacks of pneumonia and thrush (oral infection usually due to Candida albicans) since birth. The major findings of a thorough workup were very low levels of circulating lymphocytes (ie, severe lymphopenia) and low levels of circulating immunoglobulins. The attending pediatrician suspected SCID.
Laboratory Findings
Analysis of a sample of red blood cells revealed a very low activity of ADA and also a very high level (about 50 times normal) of dATP. This confirmed the diagnosis of SCID due to deficiency of ADA, the enzyme that converts adenosine to inosine (Chapter 33):
![]()
Treatment
Appropriate antibiotic treatment was commenced and the child was given periodic injections of immune globulin. In addition, she was started on weekly intramuscular injections of bovine ADA conjugated to polyethylene glycol. Bovine ADA is relatively nonimmunogenic, and conjugation to polyethylene glycol prolongs its half-life. It has been shown to be beneficial in the treatment of ADA deficiency. Her parents were informed that bone marrow transplantation was the most appropriate therapy, but the treatment was declined. In view of reports of successes with ADA gene therapy, this treatment was offered, and the parents consented. The treatment was approved by the hospital Ethics Committee. Lymphocytes and mononuclear cells were isolated from her blood using a gradient of Ficoll (a neutral highly branched polysaccharide). They were then grown in the presence of interleukin-2 (to stimulate cell division) and infected with a modified retrovirus containing inserts encoding ADA and also a gene (the NeoR gene) encoding an enzyme governing neomycin resistance, which was used to show that gene transfer had been achieved. An alternative at the present time would be to use bone marrow stem cells (which have been reported to give good increases of both B and T cells), but these were not available at the time of treatment. The autologous gene-treated cells were then injected intravenously. The child received similar injections once a month over the next year, and in addition continued to receive polyethylene glycol-conjugated ADA. Measurement of the activity of ADA revealed a sustained elevation (about 20% of normal) of the enzyme in circulating lymphocytes after 6 months of treatment; analyses using the PCR technique with NeoR probes revealed that approximately the same percentage of lymphocytes contained inserted genetic material.
Discussion
Deficiency of the activity of ADA is inherited as an autosomal recessive condition. It accounts for approximately 15% of cases of SCID; other causes involve mutations in a variety of genes affecting the function of cells of the immune system. Most of the mutations in the gene for ADA so far detected have been base substitutions, though deletions have also been detected. These mutations result in diminished activity or stability of ADA. The block of activity of ADA results in accumulation of adenosine, which in turn results in accumulation of deoxyadenosine and dATP. Elevated levels of the latter are toxic, particularly to T lymphocytes, which normally exhibit a high activity of ADA. Thus, lymphocytes are injured or killed, resulting in impairment of both cell and humoral immunity, because impairment of T-cell function can secondarily affect B-cell function.
Adenosine deaminase deficiency has become quite prominent because it is the first disease to be treated by somatic cell gene therapy. Several patients have been treated by protocols similar to that described above, which is an example of ex vivo (the lymphocytes and mononuclear cells were removed from the body prior to insertion of the ADA gene) gene therapy. One reason for selecting ADA deficiency as a suitable condition for somatic gene therapy was that cells that express the gene for ADA would have a selective advantage for growth over uncorrected cells. Gene therapy is discussed briefly in Chapter 39. Important points concerning gene therapy include that the level of expression of the affected protein should be sufficient to sustain normal function, that ideally the inserted gene should show normal regulation, and that no significant undesirable side effects should occur (eg, cancer due to insertional mutagenesis). In relation to the last point, the gene that is being delivered may insert into a gene that is essential for normal cell growth, and if this occurs that may result in the cell becoming cancerous (an example of insertional mutagenesis), as has been noted in some instances of gene therapy.
A simplified scheme of the events involved in the causation of ADA deficiency is given in Figure 57–1. Safe and effective treatment of ADA deficiency by gene therapy has been reported recently (see References).
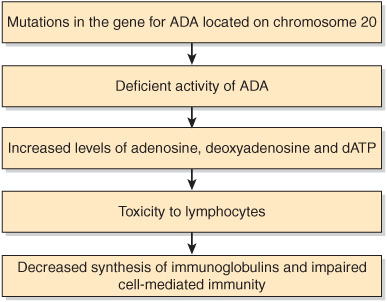
FIGURE 57–1 Summary of probable events in the causation of SCID due to ADA deficiency (OMIM 102700).
CASE 2: ALZHEIMER DISEASE (AD)
Before studying this case, the reader is advised to consult the material in Chapter 5 on AD.
Causation
Deposition of amyloid β peptide (Aβ42) in certain parts of the brain is believed by many neuroscientists to be one major cause of AD. It is thought that this 42-amino-acid peptide, occurring as beta sheets, oligomerizes and is deposited around neurons; the oligomers may be toxic to neurones. Deposition of Aβ42 may be due to excessive formation or decreased removal of the peptide. In certain cases of familial AD, specific genes have been identified (eg, these encoding amyloid precursor protein [APP], presenilins 1 and 2, and apolipoprotein E4), which affect the production or removal of Aβ42.
History and Physical Examination
A 72-year-old woman who lived on her own was found wandering around her neighborhood at 2 A.M. Her husband had died 3 years previously and her one son lived some distance away. The lady was confused and taken to the hospital. The son was notified and came immediately to see his mother. She was not able at the time of admission to give a clear history. The son volunteered that she had been diagnosed by a neurologist as having early AD, but had refused to go into a nursing home. She had home help during the day, and had not previously wandered out of her home. Sometimes a lady friend visited and spent the night at her house. In fact, she had appeared relatively normal prior to the present situation, as her son spoke to her on the phone every day of the week. However, her short-term memory had become worse in recent months, and he had become concerned about her. She was on a medication (donepezil) for AD. Otherwise she had no significant medical history. She was kept in the hospital for a couple of days, during which time her family doctor and the neurologist were consulted.
Treatment
There is no specific treatment for AD at the present time. Donepezil and several other drugs that are used in the management of AD inhibit the activity of cholinesterase, an enzyme that hydrolyzes acetylcholine (ACh) to acetate and choline. They are used because some studies had shown lower than normal levels of ACh in specimens of brains from subjects who had died of AD. They appear to produce a modest improvement of brain function and memory in some patients. Memantine, a drug that antagonizes N-methyl-D-aspartate receptors, may cause some slowing of the progression of AD. Symptoms such as depression, agitation, anxiety, and insomnia can be treated by appropriate drugs and antipsychotics may be required if psychosis intervenes. A possible preventive role of omega-3 fatty acids is under study. However, overall, there is as yet no effective therapy for AD. This patient was kept on donepezil and admitted to a nursing home that specialized in caring for patients with AD. Apart from high-quality basic care, the nursing home offered a variety of programs, including music therapy and exercise programs.
Discussion
AD is a progressive neurodegenerative condition in which decline of general cognitive function occurs, usually accompanied by affective and behavioral disturbances. At least 2 million people in the United States suffer from AD, and its prevalence is likely to increase as more people live longer. Some cases have a familial (genetic) basis, but the great majority (~90%) appears to be sporadic. AD is the most common cause of dementia, which can be defined as a progressive decline in intellectual functions, due to an organic cause, that interferes substantially with an individual’s activities. AD places a tremendous burden on families and on the health care system, as, sooner or later, most patients cannot look after themselves. The usual age at onset is over 65 years, but the disease can have an early onset (eg, in the 40s), particularly in cases where there is a genetic predisposition (see below). Survival ranges from 2 to 20 years. It is estimated that about 40% of people over 85 years of age have variable degrees of AD. Loss of short-term memory is often the first sign. AD usually progresses inexorably, and many patients are eventually completely incapacitated.
The diagnosis is usually one of exclusion. A complete neurologic exam is necessary and also a recognized mental status exam. Other forms of dementia (Lewy body, vascular, etc) must be excluded, as must other organic and psychiatric problems; various lab tests may thus be indicated to do this. In certain cases an MRI or CT scan may be indicated; these will usually reveal variable degrees of cortical atrophy and enlargement of ventricles if AD is present. Considerable research is underway to develop laboratory tests (eg, on blood or cerebrospinal fluid) that will assist in making the unequivocal diagnosis of AD.
The basic pathologic picture is of a degenerative process characterized by the death and consequent loss of cells in certain areas of the brain (eg, the cortex, hippocampus and certain other sites). Apoptosis (a programmed type of cell death in which various mechanisms, particularly the activities of proteolytic enzyme known as caspases, are activated within a cell leading to rapid cell death, see Chapter 55) may be involved in the cell death occurring in AD. At the microscopic level, neuritic plaques containing aggregated amyloid β peptide (Aβ, a peptide of 42 amino acids, occurring in beta sheets) surrounded by nerve cells containing neurofibrillary tangles (paired helical filaments formed from a hyperphosphorylated form of the microtubule associated protein, tau) are hallmarks. Deposits of Aβ2 are frequent in small blood vessels.
Intensive research is under way to determine the cause of AD. Particular interest has focused on the presence of Aβ42, the major constituent of the plaques found in AD. The term “amyloid” refers to a group of diverse extracellular protein deposits found in many different diseases (see Chapter 50). Amyloid proteins usually stain blue with iodine, like starch, which accounts for the name (amylo denotes starch). The amyloid cascade hypothesis proposes that deposition of Aβ42 is the cause of the pathologic changes observed in the brains of the victims of AD and that other changes, such as neurofibrillary tangles and vascular alterations, are secondary. Aβ42 is derived from a larger precursor protein named amyloid precursor protein (APP), whose gene is located on chromosome 21 close to the area affected in Down syndrome (trisomy 21). Individuals with Down syndrome who survive to age 50 often suffer from AD.
As shown in Figure 57–2, APP is a transmembrane protein that can be cleaved by proteases known as secretases. In step 1, APP is cleaved by either β-secretase or α-secretase to produce small nontoxic products. Then in step 2, cleavage of the β-secretase product by γ-secretase results in either the toxic Aβ42 (containing 42 amino acids) or the nontoxic Aβ40 peptide. Cleavage of the α-secretase product by γ-secretase produces the nontoxic P3 peptide. When split off from its parent protein, Aβ42 forms an insoluble extracellular deposit. Aggregation of Aβ42, produced by its oligomerization and formation of beta sheets, is thought by some to be a key event in causing AD. Recent studies have suggested that impairment of clearance of Aβ42 may be an important part of the problem in AD.
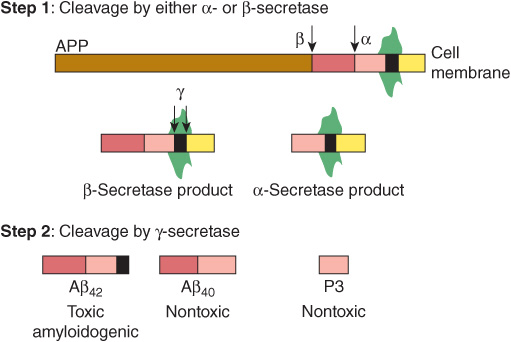
FIGURE 57–2 Simplified scheme of the formation of Aβ42. Amyloid precursor protein (APP) is digested by β-, α-, and γ-secretases. A key initial step (Step 1) is the digestion by either β-secretase or α-secretase, producing smaller nontoxic products. Cleavage of the β-secretase product by γ-secretase (Step 2) results in either the toxic Aβ42 (containing 42 amino acids) or the nontoxic Aβ40 peptide. Cleavage of the α-secretase product by γ-secretase produces the nontoxic P3 peptide. Excess production of Aβ42is a key initiator of cellular damage in Alzheimer disease (AD). Among research efforts on AD have been attempts to develop therapies to reduce accumulation of Aβ42 by inhibiting β- or γ-secretases, promoting α-secretase activity or clearing Aβ42 by use specific antibodies. (Reproduced, with permission, from Fauci AS et al [editors,] Harrison’s Principles of Internal Medicine, 17th ed, McGraw-Hill, 2008, p. 2542.)
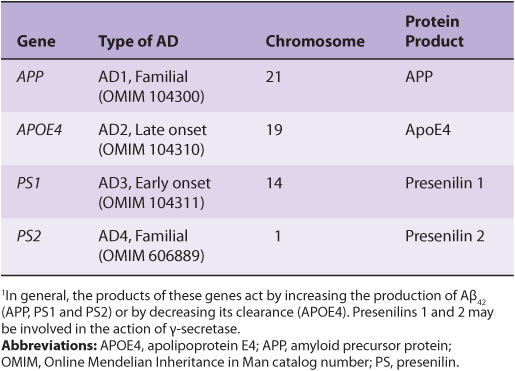
Mutations in certain genes have been found in some patients with AD (familial AD). These mutations often predispose to early onset AD. One of these genes is that encoding APP. Table 57-1 summarizes some aspects of the principal genes so far discovered. In general, the effects of the products of these genes are to enhance deposition of amyloid or to diminish its removal. Precise dissection of their mechanisms of action is underway.
TABLE 57–1 Some Genes Involved in Familial Types of Alzheimer Disease (AD)1
A second part of the amyloid cascade hypothesis is that Aβ or Aβ-containing fragments are directly or indirectly neurotoxic. There is evidence that exposure of neurons to Aβ can increase their intracellular concentrations of Ca2+. Some protein kinases, including that involved in phosphorylation of tau, are regulated by levels of Ca2+. Thus, the increase of Ca2+ may lead to hyperphosphorylation of tau and formation of the paired helical filaments present in the neurofibrillary tangles. Interference with synaptic function is also probable, perhaps secondary to neuronal damage.
Further research may reveal unexpected developments that alter the validity of the amyloid cascade theory as presented above.
Work on AD has shown the probable importance of an abnormally folded peptide in the causation of this important brain disease. It is hoped that further research on AD may result in drugs that will prevent, arrest, or even reverse AD. For example, it may be possible to develop small molecules that prevent formation or deposition of Aβ42, prevent its aggregation, or accelerate its clearance. In addition, it is possible that specific antibodies to Aβ42 or tau could prevent their putative toxic actions.
AD is one of the so-called conformational diseases (Chapters 46 and 50), in which abnormally folded proteins play a central role in the causation of a disease. Other examples of these diseases are cystic fibrosis (this chapter), alpha1 -antitrypsin disease (Chapter 50), and the prion diseases (Chapter 5).
The study of various neurodegenerative diseases is providing dramatic evidence of the importance of protein structure and function in their causation. For example, abnormal forms of the protein huntingtin play an important role in Huntington disease, abnormalities of α-synuclein play a role in some cases of Parkinson disease, and prions have been found to play key roles in the causation of bovine spongiform encephalopathy (BSE) and Creutzfeldt-Jacob disease. The application of genomic and proteomic techniques is also beginning to throw light on the causation of major psychiatric disorders, such as bipolar disease and schizophrenia. The importance of genetic and biochemical approaches in understanding disease processes has never been more clear. A simplified scheme of the causation of AD is shown in Figure 57–3.

FIGURE 57–3 A tentative scheme of the possible sequence of events in at least certain cases of AD.
CASE 3: CHOLERA
Causation
Infection by Vibrio cholerae.
History and Physical Examination
A 21-year-old female medical student working in a developing country suddenly began to pass profuse watery stools almost continuously. She soon started to vomit, her general condition declined abruptly, and she was rushed to the local village hospital. On admission, she was cyanotic, skin turgor was poor, blood pressure was 70/50 mmHg (normal 120/80 mmHg), and her pulse was rapid and weak. The doctor on duty diagnosed cholera, took a stool sample, and started treatment immediately.
Treatment
Treatment consisted of intravenous administration of a solution made up in the hospital containing 5 g NaCl, 4 g NaHCO3, and 1 g KCl per liter of pyrogen-free distilled water. This solution was initially given rapidly (100 mL per h) until her blood pressure and pulse rate were normalized. She was also given the antibiotic doxycycline. On the second day, she was able to take the oral rehydration solution recommended by the World Health Organization (WHO) for the treatment of cholera, consisting of 20 g of glucose, 3.5 g of NaCl, trisodium citrate, and dihydrate 2.9 g (or 2.5 g NaHCO3), and 1.5 g KCl per liter of drinking water.
She took amounts moderately exceeding the volume of her daily stools. Solid food was reinstituted on the fourth day after admission. She continued to recover rapidly and was discharged 7 days after admission.
Discussion
Cholera is an important infectious disease endemic in certain Asian countries and other parts of the world. Fecal contamination of water and food is the principal method of transmission. It is due to Vibrio cholerae, a bacterium that secretes a protein enterotoxin. The toxin is actually encoded by a bacteriophage (CTXϕ) resident in V cholerae. The enterotoxin is made up of one A subunit (composed of one A1 and one A2 peptide joined by a disulfide link) and five B subunits and has a molecular mass of approximately 84 kDa. In the small intestine, the toxin attaches by means of the B subunits binding to the ganglioside GM1 (Figure 15–17) present in the plasma membrane of mucosal cells (Figure 57–4). The A subunit then dissociates, and the A1 peptide passes across to the inner aspect of the plasma membrane. It catalyzes the ADP-ribosylation (using NAD+ as donor) of the GTP-binding regulatory component (Gs) of adenylate cyclase, upregulating the activity of this enzyme. Thus, adenylyl cyclase becomes chronically activated (Chapter 42). This results in an elevation of cAMP, which activates protein kinase A (PKA). This in turn via phosphorylation of CFTR and of a Na+-H+ exchanger leads to inhibition of absorption of Na+ and enhancement of secretion of Cl–. Thus, massive amounts of NaCl accumulate inside the lumen of the intestine, attracting water by osmosis and contributing to the liquid stools characteristic of cholera.
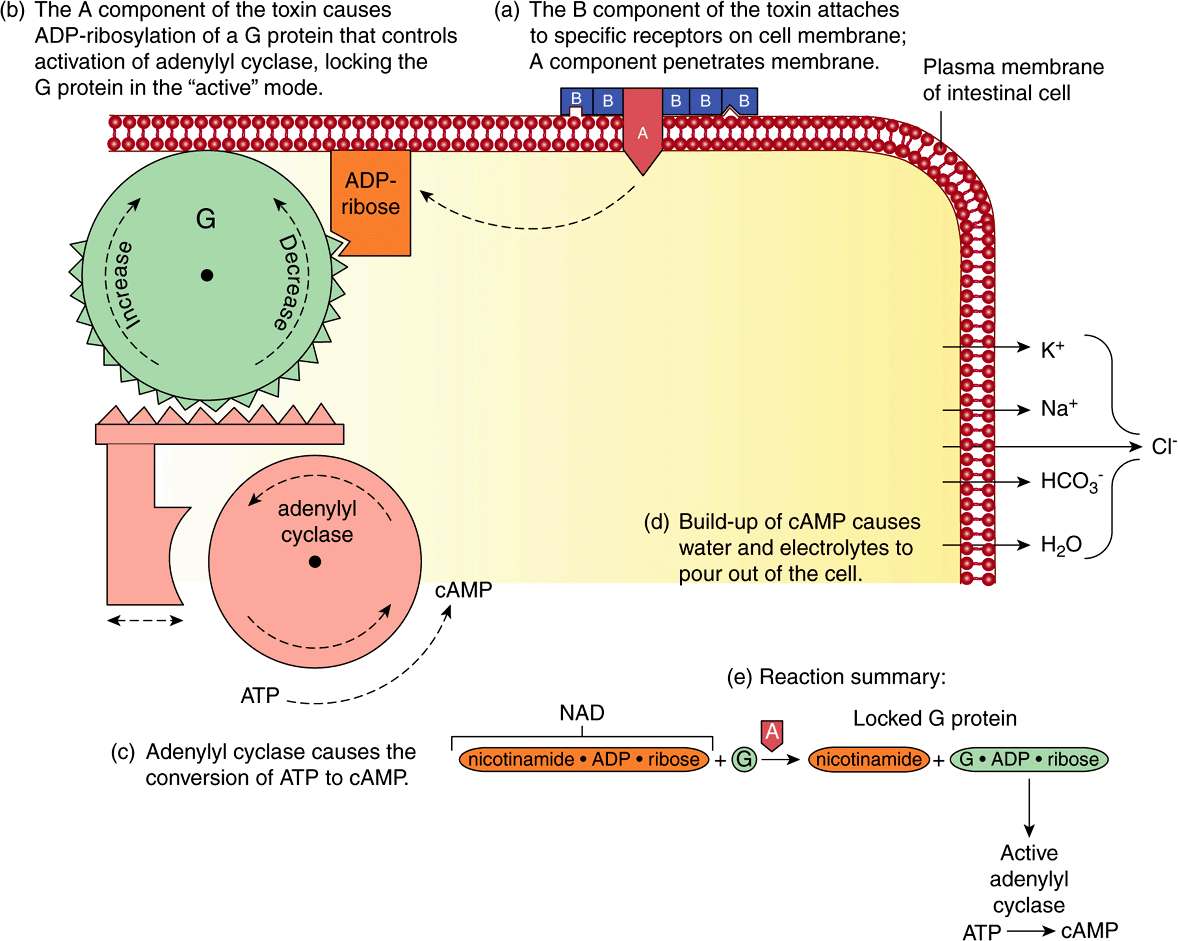
FIGURE 57–4 Diagrammatic representation of the mechanism of action of cholera toxin (CT). The toxin attaches to the plasma membrane via interaction of its B subunits with the ganglioside GM1. The A subunit crosses the membrane and catalyzes the addition of the ADP-ribose component of NAD+ to the G protein involved in stimulating adenylyl cyclase (NAD+ → ADP-ribose + nicotinamide). The addition of ADP-ribose to the G protein locks adenylyl cyclase in its active conformation, increasing the intracellular level of cAMP. This leads to phosphorylation of several membrane transporters, in turn resulting in accumulation of the ions shown and of water in the intestinal lumen, thus producing often massive diarrhea. (Reproduced, with permission, from Nester EW et al, Microbiology: A Human Perspective, 5th ed. McGraw Hill, 2007.)
Cholera toxin may also affect other molecules involved in intestinal secretion (eg, prostaglandins and nerve histamine receptors). The histologic structure of the small intestine remains remarkably unaffected, despite the loss of large amounts of Na+, Cl– water, HCO3, and K+. It is the loss of these constituents that results in the marked fluid loss (dehydration), low blood volume, acidosis, and K+ depletion found in serious cases of cholera, and which can prove fatal unless appropriate replacement therapy (as described above) is begun immediately. A person suffering from cholera can lose as much as 1 L of fluid per hour.
The recognition and easy availability of appropriate replacement fluids, such as oral rehydration solution, has led to tremendous improvement in the treatment of cholera. It should be emphasized that glucose is an essential component of oral rehydration solution (Chapter 40). Cholera toxin inhibits absorption of Na+ by intestinal cells, but it does not inhibit glucose-facilitated Na+ transport into these cells, so glucose will be absorbed and used to supply energy.
Figure 57–5 summarizes the mechanisms involved in the causation of the diarrhea of cholera.
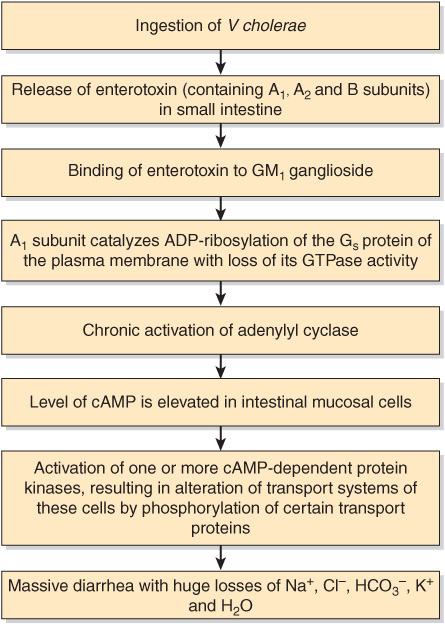
FIGURE 57–5 Summary of mechanisms involved in the causation of the diarrhea of cholera.
CASE 4: COLORECTAL CANCER
Causation
Environmental and genetic. Most if not all cancers are thought to originate by the occurrence of mutations in key genes regulating cell growth (oncogenes and tumor suppressor genes, see Chapter 55) The mutations may be inherited, but much more often various environmental influences (eg chemicals, radiation, and some viruses) are involved.
History, Physical Examination, and Results of Tests
A 62-year-old male consulted his family physician. He had noted that he had passed some fresh bright red blood in his stools several times in the previous 3 months, which he attributed to hemorrhoids. Over the previous 12 months his appetite had decreased and he had lost over 10 lbs. He had always been in good health until the past year, and was not on any medications. He had no other complaints.
In view of the history of rectal bleeding, weight loss, and the patient’s age, his physician suspected that he might have colorectal cancer and requested the patient to submit three consecutive daily specimens of feces for the fecal occult blood test. Shortly thereafter the physician received a report indicating that the results were positive. He also ordered a complete blood count and estimations of levels of serum iron, iron-binding capacity, and ferritin. The results showed a microcytic anemia (see Chapter 52), often found in patients with colorectal cancer because of bleeding from the tumor. A rectal examination was negative. No abnormalities were noted in chest x-rays.
The physician arranged a consult 4 days later with a gastroenterologist. Colonoscopy was performed 1 week later. This revealed the presence of a moderately large tumor (approximately 5 × 6 cm) in the middle of the transverse colon. Measurement of carcinoembryonic antigen (CEA), a biomarker for colorectal cancer (see below for further comments and also Chapter 7), was ordered. It was elevated (20 μg/L, normal 0-3 μg/L). Surgery was scheduled 2 weeks later, when the tumor was resected and end-to-end anastomosis performed. The regional lymph nodes were also excised and submitted along with the tumor specimen to the pathology lab. No local invasion by the tumor was noted, and no tumor was visible elsewhere in the abdomen, including the liver. The subsequent pathology report described the tumor as a relatively well-differentiated adenocarcinoma, invading the muscular mucosa. No tumor cells were noted in the lymph glands; no distant metastases were noted at the time of surgery. The TNM stage was T1N0M0 (cancer limited to the mucosa and submucosa, with an approximate 5-year survival rate of >90% [T = tumor, N = nodes, M = metastases]). (The interested reader should check the staging of tumors of the large intestine in a textbook of pathology.) In view of these findings, no chemotherapy or radiation therapy was considered necessary. Determination of CEA several weeks after surgery showed it had declined to normal levels. The patient was advised to return for follow-up at regular intervals, when, among other tests, samples of blood for measurements of CEA were taken; they remained at normal levels. A follow-up colonoscopy was performed 3 years after the operation; no tumor was seen in the colon. The patient was alive and well at 5 years after operation.
Discussion
Many aspects of the biochemistry of cancer are discussed in Chapter 55. Figures 55-1 and 55-2 summarize important features exhibited by cancer cells. Here we shall focus on a few specific aspects of colorectal cancer and also on the use of CEA as a tumor marker.
Colorectal cancer is the second most common cancer in the United States, lung cancer being number one. It can occur anywhere in the large intestine, although the rectum is the most common site. Some 95% of malignant tumors in the large intestine are adenocarcinomas (cancers of epithelial origin arising from glandular structures). About 10% of colorectal cancers occur in the transverse colon. In this case, although the tumor was moderately large, no extension from the primary site of the tumor occurred, no local nodes were involved, and no distant metastases had occurred. This was fortunate for the patient, as it meant there was an excellent prognosis and also he did not have to be subjected to chemotherapy or radiotherapy.
Most colorectal adenocarcinomas originate from adenomatous polyps. A polyp is a growth, usually benign, protruding from a mucous membrane. There are a variety of types. The one of interest here is the adenomatous polyp. Most tumors of the colon arise from such polyps although the majority of such polyps do not progress to cancer.
There are a number of well-defined genetic syndromes that predispose to colorectal cancer. The most common is hereditary nonpolyposis colorectal cancer (HNPCC), in which mutations in various genes involved in DNA mismatch repair are involved (see Chapter 35). Another relatively rare condition is familial adenomatous polyposis (adenomatous polyposis coli, APC) in which hundreds or thousands of polyps appear in the colon and rectum. The APC gene is located on chromosome 5q21 and many mutations have been described. Overall, it has been estimated that approximately 20% of colorectal cancers have a genetic basis.
Various environmental factors have been proposed as being involved in the causation of colorectal cancer. These include diets rich in saturated fat, high in calories, low in calcium, and low in fiber. How exactly each of these proposed factors operates is the subject of ongoing research. For example, dietary fat appears to enhance the production of cholesterol and bile acids by the liver. When bile acids are excreted into the bowel, bacterial enzymes may act on them to convert them to secondary bile acids, which are thought to be tumor promoters. A tumor promoter is a molecule that along with an initiator (ie a molecule that causes a mutation in DNA) leads a cell to become cancerous. Inflammatory bowel disease (eg, ulcerative colitis) is another predisposing factor to colorectal cancer.
The pioneering work of Vogelstein and colleagues on the sequence of changes occurring in tumor suppressor genes and oncogenes in dysplastic epithelium, adenomatous polyps and adenocarcinomas of the colon was described in Chapter 55 (see Figure 55–10 and Table 55-9). The reader is advised to read the appropriate section in that Chapter.
The use of tumor markers in the management of cancer was discussed in Chapter 55. CEA was one of the tumor markers described (see Table 55-15). It is a glycoprotein present in the plasma membranes of many cells. It is released into the plasma in a number of conditions, in which it can be measured by a radioimmunoassay. Levels of CEA are increased in serum in colorectal cancer, but also in other alimentary and nonalimentary cancers, and in certain noncancerous conditions.
Less than 50% of individuals with localized colorectal cancer have elevated levels of CEA, so it is not a useful screening test for this condition. Its main use is to monitor the effects of treatment and to detect recurrence of cancers such as colorectal cancer, by following changes in its level. In the present case, the level of CEA remained low for 5 years after surgery, suggesting no recurrence of cancer had occurred. A goal (probably unattainable) of research in this area would be to develop highly specific biomarkers for very early colorectal cancer and for other very early tumors that would be positive in 100% of cases and negative in 100% of normal individuals!
Figure 57–6 summarizes some major factors leading to the development of colorectal cancer.
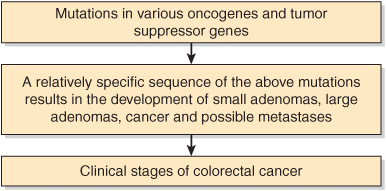
FIGURE 57–6 Simplified scheme of the multistep causation of colorectal cancer.
CASE 5: CYSTIC FIBROSIS (CF)
Causation
Genetic (mutations in the gene encoding the cystic fibrosis transmembrane regulator [CFTR] protein).
History and Physical Examination
A 1-year-old girl, an only child of Caucasian background, was brought to the clinic at the Hospital for Sick Children by her mother. She had been feverish for the past 24 hours and was coughing frequently. The mother stated that her daughter had experienced three attacks of “bronchitis” since birth, each of which had been treated with antibiotics by their family physician. The mother had also noted that her daughter had been passing somewhat bulky, foul-smelling stools for the past several months and was not gaining weight as expected. In view of the history of pulmonary and gastrointestinal problems, the attending physician suspected that the patient might have CF although no family history of this condition was elicited.
Laboratory Findings
Chest x-rays showed signs consistent with bronchopneumonia. Culture of sputum revealed predominantly Pseudomonas aeruginosa. Fecal fat was increased. A quantitative pilocarpine iontophoresis sweat test was performed, and the sweat Cl– was 70 mmol/L (>60 mmol/L is abnormal); the test was repeated a week later with similar results.
Treatment
The child was given an appropriate antibiotic and referred to the cystic fibrosis clinic for further care. A comprehensive program was instituted to look after all aspects of her health, including psychosocial considerations. She was started on a pancreatic enzyme preparation (given with each meal) and placed on a high-calorie diet supplemented with multivitamins and vitamin E. Postural drainage was begun for the thick pulmonary secretions. Subsequent infections were treated promptly with appropriate antibiotics and with an aerosolized recombinant preparation of human DNase that digests the DNA of microorganisms present in the respiratory tract. At age 6 years, she had grown normally, had been relatively free of infection for a year, was attending school, and making satisfactory progress. Serious chronic cases of CF in which the lungs are severely compromised are candidates for lung transplants, although the efficacy of this treatment has been challenged recently.
Research on gene therapy for CF is under examination (eg, using recombinant viruses encoding the CFTR protein). Another line of research is investigating whether small molecules can be found for clinical use that help abnormally folded CFTR molecules re-fold into at least partially active molecules.
Discussion
CF is a prevalent and usually serious genetic disease among whites in North America. It affects approximately 1:2500 individuals and is inherited as an autosomal recessive disease; about 1 person in 25 is a carrier. It is a disease of the exocrine glands, with the respiratory and gastrointestinal tracts being most affected. A diagnostic hallmark is the presence of high amounts of NaCl in sweat, reflecting an underlying abnormality in exocrine gland function (see below). Pilocarpine iontophoresis has generally been used to allow collection of sufficient amounts of sweat for analysis. Iontophoresis is a process by which drugs are introduced into the body (in this case the skin) via use of an electrical current. Its use is diminishing as the availability of specific genetic probes increases.
The classic presentation of CF is that of a young child with a history of recurrent pulmonary infection and signs of exocrine insufficiency (eg, fatty, bulky stools due to a lack of pancreatic lipase), as in the present case. However, the disease is clinically heterogeneous, which at least partly reflects heterogeneity at the molecular level (see below). Approximately 15% of patients may have sufficient pancreatic function to be classified as “pancreatic sufficient.”
For reasons related to abnormalities in Cl– and Na+ transport (see below), the pancreatic ducts and the ducts of certain other exocrine glands become filled with viscous mucus, which leads to their obstruction. This mucus is also present in the bronchioles, leading to their obstruction; this favors the growth of certain bacteria (eg, Staphylococcus aureus and Pseudomonas aeruginosa) that cause recurrent bronchopulmonary infections, eventually seriously compromising lung function. In turn, the pulmonary disease can lead to right ventricular hypertrophy and possible heart failure. Patients usually die of pulmonary infection or heart failure. In recent years, more patients have been living into their 30s and later, as the condition is now diagnosed earlier and appropriate comprehensive therapy started. Sometimes, problems due to lack of pancreatic secretions can be present at birth, the infants presenting with intestinal obstruction due to very thick meconium (meconium ileus). Other patients, less severely affected, may not be diagnosed until they are in their teens or later. CF also affects the genital tract and most males and many females are infertile.
In 1989, results of a collaborative program between Canadian and American scientists revealed the nature of the genetic lesion in the majority of sufferers from CF. The gene involved in CF was the first to be cloned solely on its position determined by linkage analysis (positional cloning) and constituted an enormous amount of painstaking labor and a tremendous triumph for “reverse genetics”. By reverse genetics is meant that the gene was isolated based on its map location, and not with the availability of chromosomal rearrangements or deletions (in contrast to, for example, the isolation of the gene involved in Duchenne muscular dystrophy). The success of this Herculean effort showed that, at least in theory, the molecular basis of any genetic disease could be revealed by similar approaches. More recent advances (eg, outcomes of the Human Genome Project) have further facilitated identification of “disease genes.”
Table 57-2 summarizes the major strategies used in detecting the gene involved in the causation of CF. The protein product of the gene encodes an integral membrane protein of approximately 170 kDa. It was named CFTR and was found to be a cAMP-responsive chloride transporter, helping to explain the high chloride content of sweat found in patients with CF.
TABLE 57–2 Some Strategies Used in Isolating the Gene Involved in CF

Some of the characteristics of the gene and of the CFTR protein are listed in Table 57-3. The major mutation initially located in the gene was deletion of three bases encoding phenylalanine residue 508 (ΔF508); in North America, approximately 70% of CF carriers have this mutation. Subsequent work has revealed well over 1000 different mutations in the gene. A variety of different types of mutations have been found, including small deletions, insertions, and missense and nonsense mutations. Because of the importance of early diagnosis, in certain countries, all newborn infants are now undergoing genetic screening for CF. Elsewhere, techniques to detect deletion of ΔF508 and a number of the other most frequent mutations are being used to confirm the diagnosis of CF, to detect carriers and in prenatal diagnosis.
TABLE 57–3 Some Characteristics of the Gene for the CFTR Protein and of the Protein Itself

The CFTR protein (Figure 57–7) consists of two similar halves, each containing six transmembrane regions and a nucleotide (ATP)-binding fold (NBF). The two halves of the molecule are joined by a regulatory domain. F508 is located in NBF1. The protein shows similarities in structure to certain other proteins that use ATP to transport molecules across cells membranes (eg, P-glycoprotein, involved in resistance to certain cancer chemotherapeutic agents).
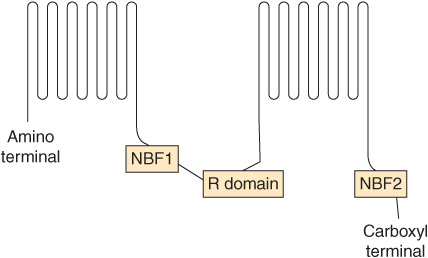
FIGURE 57–7 Diagram of the structure of the CFTR protein (not to scale). The protein contains 12 transmembrane segments, two nucleotide-binding folds or domains (NBF1 and NBF2), and one regulatory (R) domain. NBF1 and NBF2 bind ATP and couple its hydrolysis to transport of Cl–; they are called ATP-binding cassettes (ABCs), a feature found in many membrane transporters. Phe 508, the major locus of mutations in CF, is located in NBF1.
Normally, CFTR is synthesized on bound polyribosomes and exported to the plasma membrane, where it functions. Mutations can affect CFTR is a number of ways, summarized briefly in Table 57-4. Many mutations affect the folding of the protein, markedly reducing its function; this classifies CF as a conformational disease or a disease due to a deficiency in proteostasis (see Chapter 46 and the discussion of Alzheimer disease in this chapter). Mutations affect many other proteins in a similar manner to those summarized in Table 54-4, affecting their synthesis, processing, or function.
TABLE 57–4 Some Mechanisms by Which Mutations May Affect the CFTR1 Protein

Figure 57–8 summarizes some of the mechanisms involved in the causation of CF.
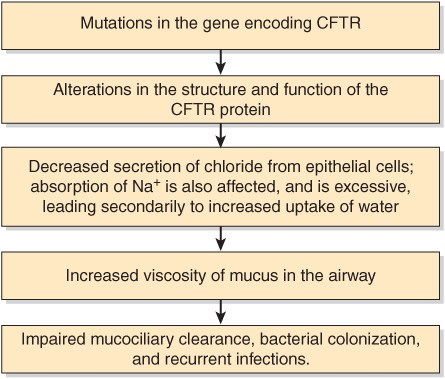
FIGURE 57–8 Summary of possible mechanisms involved in cells in the airways of individuals with cystic fibrosis (OMIM 219700) who have pulmonary pathology. In individuals of Caucasian origin, 70% of the mutations occur at one locus, resulting in deletion of ΔF508 from the CTR protein. However, over 1000 mutations have been identified in the CFTR gene. Basically, the CFTR protein acts normally as a cAMP-regulated transporter involved in secretion of Cl–, but in addition normally inhibits absorption of Na+ by a Na+ channel. The viscosity of the mucus in the pancreatic ductules is also increased, leading to their obstruction. The details of how abnormalities of CFTR affect ion transport in the pancreas are somewhat different than in the lung.
CASE 6: DIABETIC KETOACIDOSIS (DKA)
Causation
Endocrine (due to deficiency of insulin).
History and Physical Examination
A 14-year-old girl was admitted to a children’s hospital in coma. Her mother stated that the girl had been in good health until approximately 2 weeks previously, when she developed a sore throat and moderate fever. She subsequently lost her appetite and generally did not feel well. Several days before admission, she began to complain of undue thirst and also started to get up several times during the night to urinate. Her family doctor was out of town and her mother felt reluctant to contact another physician. However, on the day of admission the girl had started to vomit, had become drowsy and difficult to arouse, and accordingly had been brought to the emergency department. On examination she was dehydrated, her skin was cold, she was breathing in a deep sighing manner (Kussmaul respiration), and her breath had a fruity odor. Her blood pressure was 90/60 and her pulse rate 115/min. She could not be aroused. A diagnosis of type 1 diabetes mellitus (formerly called insulin-dependent) with resulting ketoacidosis and coma (DKA) was made by the intern on duty.
Laboratory Findings
The admitting diagnosis was confirmed by the laboratory findings, kindly supplied by Dr. ML Halperin:
Plasma or serum results (normal levels in parentheses, SI Units):
• Glucose, 50 (4.2-6.1 mmol/L)
• Ketoacids ++++ (trace)
• Bicarbonate, 6 (22-30 mmol/L)
• Urea nitrogen, 15 (2.5-7.1 mmol/L)
• Arterial blood pH, 7.07 (7.35-7.45)
• Na+, 136 (136-146 mmol/L)
• Cl–, 100 (102-109 mmol/L)
• pCO2 2.7 (4.3-6.0 kPa [or 32-45 mmHg])
• Anion gap, 31 (7-16 mmol/L) (The anion gap is calculated from plasma ![]() .)
.)
• Potassium, 5.5 (3.5-5.0 mmol/L)
• Creatinine, 200 (44-80 μmol/L)
• Albumin 50 (41-53 g/L)
• Osmolality, 325 (275-295 mOsm/kg serum water)
• Hematocrit, 0.500 (0.354-0.444)
Urine results:
• Glucose, ++++ (normal -)
• Ketoacids, ++++ (normal -)
Treatment
The most important initial measures in treatment of diabetes ketoacidosis are intravenous administration of insulin and saline solution. This patient was given intravenous insulin (10 units/h) added to 0.9% NaCl. Glucose was withheld until the level of plasma glucose fell below 15 mM. Insulin and glucose facilitate entry of K+ into cells. KCl was also administered cautiously, with plasma K+ levels monitored every hour initially. Continual monitoring of K+ levels is extremely important in the management of diabetic ketoacidosis because inadequate management of K+ balance is the main cause of death. Bicarbonate is not needed routinely, but may be required if acidosis is very severe.
Discussion
The precise cause of type 1 (insulin-dependent) diabetes mellitus has not been elucidated, and is under intense investigation. Genetic, environmental and immunologic factors have all been implicated. A very tentative scheme of the chains of events is the following. Patients with this type of diabetes have a genetic susceptibility (a large number of genes, including histocompatibility genes located on chromosome 6, have been implicated), which may predispose to a viral infection (eg, by coxsackie or rubella viruses). The infection and consequent inflammatory reaction may alter the antigenicity of the surface of the pancreatic B cells and set up an autoimmune reaction involving both cytotoxic antibodies and T lymphocytes. This leads eventually to widespread destruction of beta cells, resulting in type I diabetes mellitus. Perhaps the sore throat this patient had several weeks before admission reflected the initiating viral infection.
The marked hyperglycemia, glucosuria, ketonemia, and ketonuria confirmed the diagnosis of DKA. The low pH indicated a severe acidosis due to the greatly increased production of acetoacetic acid and β-hydroxybutyric acid. The low levels of bicarbonate and pCO2 confirmed the presence of a metabolic acidosis with partial respiratory compensation (the hyperventilation). Calculation of the anion gap is useful in a number of metabolic situations. In this case it is elevated because of the presence of excess ketoacids in the blood. There are a number of other causes of elevation of the anion gap, including lactic acidosis and intoxication by methanol, ethylene glycol, and salicylates.
The elevated values of urea and creatinine indicated some renal impairment (due to diminished renal perfusion because of low blood volume secondary to dehydration), dehydration, and increased degradation of protein. A high plasma level of potassium is often found in DKA owing to a lowered uptake of potassium by cells in the absence of insulin. Thus, the clinical picture in DKA reflects the abnormalities in the metabolism of carbohydrate, lipid, and protein that occur when plasma levels of insulin are sharply reduced. The increased osmolality of plasma due to hyperglycemia also contributes to the development of coma in diabetic ketoacidosis. It should be apparent that the rational treatment of a patient with DKA depends on thorough familiarity with the actions of insulin.
A general scheme of the events occurring in DKA is given in Figure 57–9.
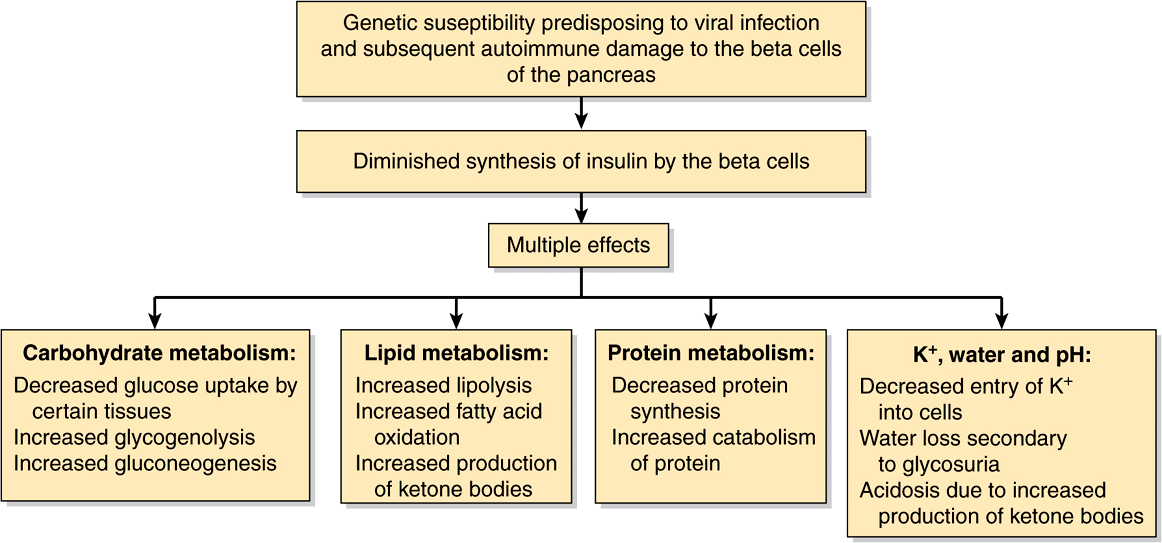
FIGURE 57–9 Summary of some mechanisms involved in the causation of the ketoacidosis of type 1 diabetes mellitus (OMIM 222100).
CASE 7: DUCHENNE MUSCULAR DYSTROPHY (DMD)
Causation
Genetic (mutations in the gene encoding the protein dystrophin).
History and Physical Examination
A 4-year-old boy was brought to a children’s hospital clinic. His mother was concerned because she had noticed that her son was walking awkwardly, fell over frequently, and had difficulty climbing stairs. There were no siblings, but the mother had a brother who died at age 19 of muscular dystrophy. The pediatrician on call noted muscular weakness in both the pelvic and shoulder girdle. Modest enlargement of the calf muscles was also noted. Because of the muscle weakness and its distribution, the pediatrician made a provisional diagnosis of DMD.
Laboratory and Other Findings
The activity in serum of creatine kinase (CK) was markedly increased. It was decided to proceed directly to mutation analysis using a sample of the patient’s lymphocytes. This showed a large deletion in the gene for dystrophin, confirming the diagnosis of DMD. This saved the patient from being tested by electromyography and also from having a muscle biopsy performed; these tests, along with Western blotting for detection of dystrophin, were routinely performed prior to the availability of mutation analysis, and still may be performed in certain circumstances.
Discussion
The family history, the typical distribution of muscular weakness, the elevation of the activity in serum of CK, and the results of mutation analysis confirmed the provisional diagnosis of DMD. This is a severe X-linked degenerative disease of muscle. It has a prevalence of approximately 1:3500 live male births. It affects young boys, who first show loss of strength in their proximal muscles, leading to a waddling gait, difficulty in standing up, and eventually very severe weakness. Death generally occurs from respiratory insufficiency.
The cause of DMD was revealed in 1986-1987. Various studies led to localization of the defect to the middle of the short arm of the X chromosome and to subsequent identification of a segment of DNA that was deleted in patients with DMD. Using the corresponding nondeleted segment from normal individuals, a cDNA was isolated derived by reverse transcription from a transcript (mRNA) of 14 kb that was expressed in fetal and adult skeletal muscle. This was cloned and the protein product was identified as dystrophin, a 400-kDa red-shaped protein of 3685 amino acids. Dystrophin was absent or markedly reduced in electrophoretograms of extracts of muscle from patients with DMD and from mice with an X-linked muscular dystrophy. Antibodies against dystrophin were used to study its localization in muscle; it is associated with the sarcolemma (plasma membrane) of normal muscle and was absent or markedly deficient in patients with DMD. A less severe reduction in the amount of dystrophin, or a reduction in its size, is the cause of Becker muscular dystrophy, a milder type of muscular dystrophy. Whereas the gene deletions and duplications found in DMD tend to cause frame-shift mutations, the same types of mutations in Becker MD are generally in-frame, and thus synthesis of dystrophin is not as affected in the latter.
Dystrophin appears to have four domains, two of which are similar to domains present in α-actinin (another muscle protein) and one to a domain in spectrin, a protein of the cytoskeleton of the red blood cell. As shown in Figure 49–11, dystrophin interacts with actin, syntrophin, and β-dystroglycan. Its function is not clear, but it may play a role in transmembrane signaling and in stabilizing the cytoskeleton and sarcolemma.
Deficiency of dystrophin may affect the integrity of the sarcolemma, resulting in increased osmotic fragility of dystrophic muscle or permitting excessive influx of Ca2+. The gene coding for dystrophin is the largest human gene recognized to date (~2500 kb, 79 exons), which helps explain the observation that approximately one third of cases of DMD are new mutations. Attempts are being made to produce dystrophin by recombinant DNA technology and perhaps eventually administer it to patients. The availability of probes for dystrophin facilitates prenatal diagnosis of DMD by chorionic villus sampling or amniocentesis. The demonstration of lack of dystrophin as a cause of DMD has been one of the major accomplishments of the application of molecular biology to the study of human diseases.
There are many different types of muscular dystrophy. The molecular causes of many of them have been elucidated. Not surprisingly, perhaps, they have been found to be due to a variety of mutations in genes that encode specific muscle proteins, such as those shown in Figure 49–11 and others not shown. The various muscular dystrophies can be classified on the basis of their clinical features (eg, affecting the limb girdle, etc), or increasingly on the basis of the genes or proteins affected by the causative mutations. Dystrophin also occurs in heart muscle and the brain. Its occurrence in the former can result in cardiomyopathy. The absence of dystrophin in brain results in an IQ of less than 75 being observed in some 25% of boys with DMD.
Treatment
At present, no specific therapy for DMD exists. Treatment in this case was thus essentially symptomatic. He was enrolled in a special muscular dystrophy clinic, started on prednisone (which can slow down the progress of DMD for a few years) and encouraged to undertake mild exercise. A physiotherapist was available when needed. Portable respirators have proven very useful when breathing is affected. The mother was advised to seek genetic counseling regarding future pregnancies. At different times, a variety of therapeutic measures intended to benefit patients with DMD have been used. These include the use of myoblast transfer (to supply dystrophin), antisense oligonucleotides (to skip mutated dystrophin gene exons), CoE Q10 (to possibly increase muscle strength), creatine monohydrate (to perhaps help build up muscle mass), pentoxyfylline (anti-inflammatory), and gentamicin (may ignore premature stop codons in the dystrophin gene). None appears to have been very successful. A small trial using dystrophin gene transfer was scheduled to start in the United States in January 2008.
Figure 57–10 summarizes the mechanisms involved in the causation of DMD.
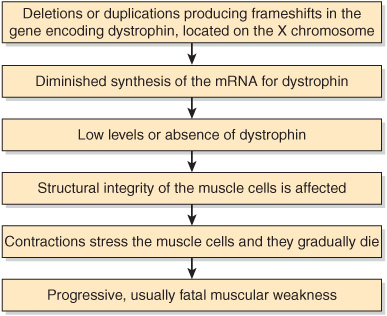
FIGURE 57–10 Summary of mechanisms involved in the causation of Duchenne muscular dystrophy (OMIM 310200).
CASE 8: ETHANOL INTOXICATION, ACUTE
Causation
Chemical (due to excess intake of ethanol).
History and Physical Examination
A 52-year-old man was admitted to the emergency department in a coma. Apparently, he had become increasingly depressed after the death of his wife 1 month previously. Before her death he had been a moderate drinker, but his consumption of alcohol had increased markedly over the last few weeks. He had also been eating poorly. His married daughter had dropped round to see him on Sunday morning and had found him unconscious on the living room couch. Two empty bottles of rye whiskey were found on the living room table. On examination, he could not be roused, his breathing was deep and noisy, alcohol could be smelled on his breath, and his temperature was 35.5°C (normal; 36.3-37.1°C). The diagnosis on admission was coma due to excessive intake of alcohol.
Laboratory Findings
The pertinent laboratory results were alcohol 500 mg/dL, glucose 2.7 mmol/L (normal: 4.2-6.1), lactate 8.0 mmol/L (normal: 0.5-1.6), and blood pH of 7.21 (normal: 7.35-7.45).
These results were consistent with the admitting diagnosis, accompanied by a metabolic acidosis.
Treatment
He was started on intravenous normal saline, and then, because of the very high level of blood alcohol and the coma, it was decided to start hemodialysis immediately. This directly eliminates the toxic ethanol from the body but is only required in very serious cases of ethanol toxicity. In this case, the level of blood alcohol fell rapidly and the patient regained consciousness later the same day. Intravenous glucose (5%) was administered after dialysis was stopped to counteract the hypoglycemia that this patient exhibited. The patient made a good recovery and was referred for psychiatric counseling.
Discussion
Excessive consumption of alcohol is a major health problem in most societies. The present case deals with the acute, toxic effects of a very large intake of ethanol. A related problem, which is not discussed here but which has many biochemical aspects, is the development of liver cirrhosis in individuals who maintain a high intake of ethanol (eg, 80 g of absolute ethanol daily for more than 10 years).
From a biochemical viewpoint, the major question concerning the present case is how does ethanol produce its diverse acute effects, including coma, lacticacidosis, and hypoglycemia? The clinical viewpoint is how best to treat this condition.
The metabolism of ethanol was described in Chapter 25; it occurs mainly in the liver and involves two routes. The first and major route uses alcohol dehydrogenase and acetalde-hyde dehydrogenase, converting ethanol via acetaldehyde to acetate (Chapter 25), which is then converted to acetyl-CoA. NADH + H+ are produced in both of these reactions. The intracellular ratio of NADH/NAD+ can thus be increased appreciably by ingestion of large amounts of ethanol. In turn, this can affect the Keq of a number of important metabolic reactions that use these two cofactors. High levels of NADH favor formation of lactate from pyruvate, accounting for the lactic acidosis. This diminishes the concentration of pyruvate (required for the pyruvate carboxylase reaction, Chapter 17) and thus inhibits gluconeogenesis. In severe cases, when liver glycogen is depleted and no longer available for glycogenolysis, hypoglycemia results. The second route involves a microsomal cytochrome P450 (microsomal ethanol oxidizing system), also producing acetaldehyde (Chapter 25). Acetaldehyde is a highly reactive molecule and can form adducts with proteins, nucleic acids, and other molecules. It appears likely that its ability to react with various molecules is involved in the causation of the toxic effects of ethanol.
Apart from the metabolic studies just mentioned, early studies suggested that ethanol produced many of its acute intoxicating effects by disordering lipids in the cell membranes of neurons. More recent studies have focused on proteins, particularly receptors and ion channels (see Chapters 40 and 41), which play important roles in the normal function of neurons and other cells. A variety of studies have revealed that ethanol inhibits, or in some cases activates various receptors and ion channels. These include GABA (γ-amino-butyric acid), nicotinic acetylcholine, glutamate, and NMDA (N-methyl-D-aspartate) receptors, and various K+ and Ca2+ channels. Alterations of the activities of receptors can affect intracellular signaling pathways, contributing to the effects of ethanol. One way in which ethanol may disturb the functions of these proteins is by replacing water molecules at various critical sites. Further research should help account for the acute effects of ethanol on mental status. Figure 57–11 summarizes some of the major mechanisms involved in the causation of toxicity by ethanol.
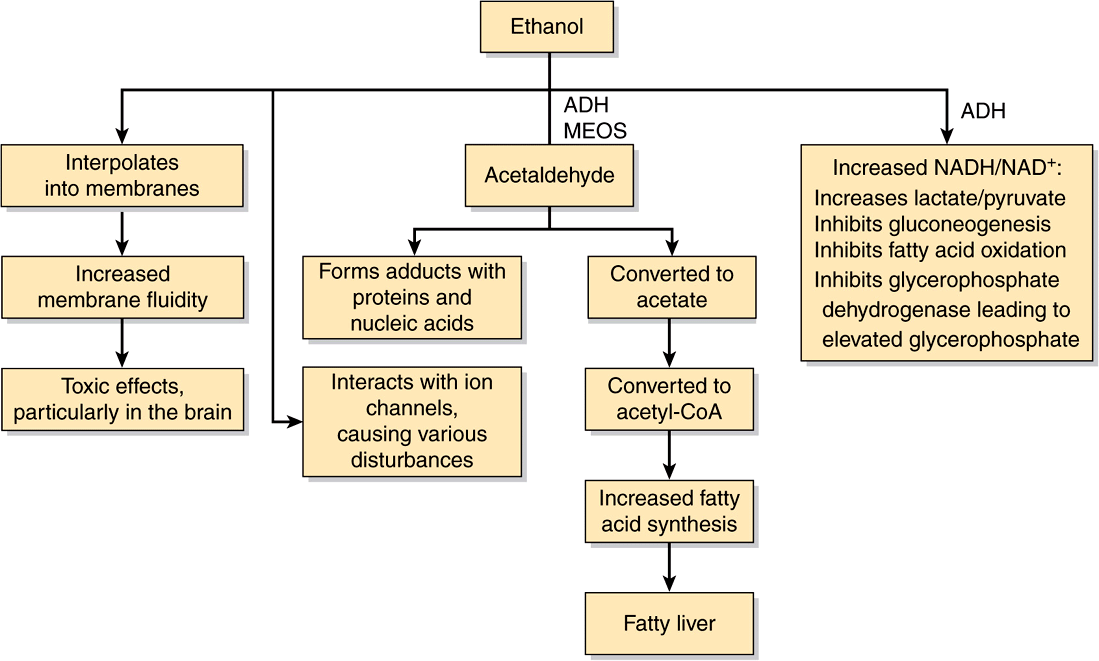
FIGURE 57–11 Summary of some mechanisms involved in acute ethanol toxicity. (ADH, alcohol dehydrogenase; MEOS, microsomal ethanol oxidizing system, involving cytochrome P450 species CYP2E1.)
CASE 9: GOUT, ACUTE
Before studying this case, the reader is advised to consult the material in Chapter 33 dealing with uric acid.
Causation
Deposition of crystals of monosodium urate (MSU) in one or more joints and various tissues. The great majority of cases (~90%) are associated with decreased renal excretion of MSU, but in certain cases, increased production of MSU is involved, and increased dietary intake of purines may play a role.
History and Physical Examination
A moderately obese 64-year-old male appeared at the emergency department complaining of severe pain of 12-hour duration in his left big toe. He stated that he regularly had at least two to three drinks of Scotch whisky every evening after work. He had no other significant medical history. On examination, his left big toe was found to be red and markedly swollen around the metacarpophalangeal joint, and exquisitely sensitive. There was no evidence of arthritis elsewhere. Because of the history and location of the affected joint, the intern on duty suspected that the patient was having an attack of acute gout. She ordered a number of lab tests, including a white cell count, determination of serum uric acid, and X-ray examination of the affected joint. The serum level of uric acid, was 0.61 mmol/L (normal, 0.18-0.41 mmol/L in males); the white cell count was at the high end of normal. The x-ray findings were nonspecific; no indication of chronic arthritis was evident. Under local anesthesia, arthrocentesis was performed on the affected joint, and a small amount of synovial fluid withdrawn and sent to the laboratory for detection of cells and crystals. Typical needle-shaped crystals of MSU showing negative birefringence were detected in the synovial fluid, as were neutrophils.
Treatment
The patient was started on a suitable dose of a nonsteroidal anti-inflammatory drug (NSAID) to relieve the acute inflammation and pain. He was referred to a rheumatologist for ongoing treatment; she continued administration of the NSAID. Several months later he had another acute attack of joint pain, this time in his right knee. His plasma level of uric acid was 0.57 mmol/L. Daily excretion of uric acid was measured and found to be ~9.0 mmol (~1500 mg) (normal 3.6-5.4 mmol/d). The rheumatologist decided to start him on long-term therapy with allopurinol, a drug used to decrease formation of uric acid by inhibiting xanthine oxidase, the enzyme responsible for formation of uric acid from xanthine (see Chapter 33). In addition, the patient was referred to a dietitian regarding losing weight, advised to drink plenty of fluids, to markedly limit his intake of alcohol, to restrict intake of purine-rich foods (eg, anchovies and red meat), and started on a regular exercise program. Until the present, the patient has had no further attacks of acute arthritis.
Discussion
Uric acid is formed from purine nucleosides (eg, adenosine and guanosine) produced by the breakdown of nucleic acids and other molecules (eg, ATP), and in humans is the end-product of purine catabolism. The daily synthesis rate is estimated to be 1.8 mmol (~300 mg), with a total body pool of approximately 7.2 mmol (1200 mg in adult males, and about one-half that in females). In individuals with gout, the total body pool can be as large as 180 mmol (30,000 mg).
The enzyme involved in formation of uric acid is xanthine oxidase (see Chapter 33). Humans do not possess the peroxisomal enzyme uricase (urate oxidase), which is involved in the degradation of uric acid to allantoin. Uricase is now available as a medication to help lower levels of uric acid. Approximately 70% of uric acid is excreted by the kidneys and the remaining by the gut. Uric acid exhibits antioxidant properties (see Chapter 45); the possible significance of this is under investigation.
Gout is a type of arthritis, acute or chronic, due to deposition of crystals of MSU usually in relatively avascular areas, such as cartilage and tissues around joints, and also where body temperature is lower (eg, the ears, distal ends of the limbs). When crystals of MSU are deposited in synovial fluid, they elicit an inflammatory reaction. In acute gout, this consists mainly of neutrophils. The inflammatory reaction causes the characteristic signs and symptoms of heat, pain, swelling, and redness experienced in acute gout. It is generally important to ascertain that the characteristic crystals of MSU are actually present in the synovial fluid of an affected joint, as other crystals (eg, calcium pyrophosphate) may cause signs and symptoms similar to gout. One joint is usually affected initially (ie, monoarticular arthritis), often the metacarpophalangeal joint of the big toe, as in this case. One factor helping to account for this is that the temperatures of the joints of the lower extremities are lower than elsewhere in the body.
MSU has a solubility value in plasma of ~0.42 mmol/L at 37°C. It is much more soluble than uric acid, which is the major ionic species below pH 5.75 (the pKa value for the dissociation of uric acid to urate). This difference is particularly important in urine, where calculi of uric acid may form at acidic values of pH. When the above concentration is exceeded, plasma is supersaturated with respect to MSU. The concentration at which precipitation of MSU in tissues and joints occurs appears to vary, for unknown reasons. Before treatments were available to prevent chronic gout occurring (eg, allopurinol, see below), large aggregates of MSU would accumulate in various tissues; these are called tophi, and can attain a considerable size. Tophi may still occur if gout is not diagnosed and treated early.
Gout is usually preceded and accompanied by hyperuricemia (plasma uric acid level >0.41 mmol/L). The sequence shown in Figure 57–12 is often involved. Chronic gout can be prevented if appropriate treatment is instituted following an attack of acute gout. Hyperuricemia is much commoner in males, although its incidence in females increases after menopause. It should be noted that approximately 30% of people experiencing an attack of gout may have normal levels of MSU in the plasma. Hyperuricemia is caused by decreased renal excretion, increased production, or increased intake of uric acid. Decreased renal excretion is involved in the great majority of cases of gout, and genetic factors are likely involved. Many kidney diseases affect renal excretion, as does acidosis caused by various metabolic conditions. A variety of drugs (eg, certain diuretics and also salicylates) interfere with excretion of uric acid. Handling of MSU by the kidneys is complex, including phases of glomerular filtration, reabsorption, secretion, and further reabsoption in various parts of the renal tubule. The precise contributions of alterations of these phases to the causation of hyperuricemia have not been clearly defined as yet. Increased production can occur due to certain enzyme abnormalities (eg, deficiency of hypoxanthine-guanine phosphoribosyl transferase [HGPRT] and overactivity of PRPP synthetase) although these are uncommon (see Chapter 33). In Lesch-Nyhan syndrome, mutations of the gene encoding HGPRT are involved (see Chapter 33), and gout can be a feature. Death of cancer cells caused by chemotherapy leads to degradation of their nucleic acids, and thus to increased formation of purines. Increased intake can occur via ingestion of purine-rich foods, such as sweetbreads and certain meats, although this is not thought to be a major contributor to elevation of serum uric acid.
![]()
FIGURE 57–12 One common sequence of events leading to chronic gout.
The role of intake of alcohol in precipitating gout has long been recognized. Ingestion of ethanol can lead to formation of lactic acid, which inhibits secretion of uric acid. In addition, ethanol appears to promote breakdown of ATP, leading to increased production of purines from which uric acid is formed. Also, the solubility of MSU is markedly diminished as the pH in tissues drops, a situation favored by increased production of lactic acid.
Renal stones (urate calculi) develop frequently in patients with chronic gout; the risks of these are lessened by therapy with allopurinol.
For treatment of the acute inflammation and pain of acute gout, an NSAID is generally used. Colchicine is also effective in blocking inflammation caused by MSU crystals. It is known to bind to free tubulin causing the depolymerization of microtubules (Chapter 49); this may prevent the movement of neutrophils into an area containing crystals of MSU. However, it may cause nausea and vomiting. A corticosteroid or ACTH may also be employed for their anti-inflammatory effects. For long-term management, intended to prevent or reverse any complications that may have arisen, allopurinol is used to chronically inhibit the production of uric acid from xanthine. Uricase may also be used in some cases.
Uricosuric drugs (which increase the rate of excretion of uric acid) may be used instead of allopurinol if under-excretion of urate is involved; these include probenecid, sulfinpyrazone, and benzbromarone. Any accompanying conditions should be addressed (eg, obesity, hypertension, hypertriglyceridemia, alcoholism, renal disease). If gout is treated early, it is compatible with a normal lifespan.
A simplified scheme of the causation of acute gout is shown in Figure 57–13.
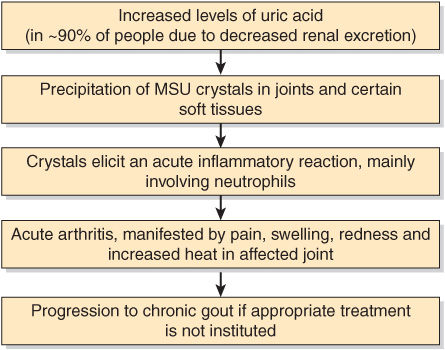
FIGURE 57–13 Simplified scheme of some of the events involved in the causation of gout.
CASE 10: HEREDITARY HEMOCHROMATOSIS
Before studying this case, the reader is advised to consult the material in Chapter 50 on iron and its metabolism.
Causation
Genetic (due to mutations in the HFE gene or certain other genes whose protein products affect the metabolism of iron).
History and Physical Examination
A 50-year-old man visited his family doctor complaining of fatigue, low libido, and moderate generalized joint pains of approximately 1-year duration. The joint pains were mostly in the fingers, wrists, hips, knees, and ankles. His parents, both dead, were born in Scotland but emigrated to Canada in early adulthood. The patient had no siblings and did not smoke or drink. He occasionally took acetaminophen for his joint pains, but otherwise was not receiving any medication. An uncle had died of liver cancer about 10 years previously. In addition to stiffness and slight swelling over some joints, the physician noted grayish skin pigmentation, most prominent in exposed parts, and for that reason referred the patients to an internist, who also noted that the liver edge was firm and palpable just below the costal margin. The internist suspected hereditary hemochromatosis and ordered appropriate laboratory tests as well as x-rays of the hands, hips, knees, and ankles.
Laboratory Findings
Normal reference values in parentheses:
• Hb, 120 g/L (133-162 g/L, males)
• RBC, 4.6 × 1012/L (4.30-5.60 × 1012/L, males)
• Glucose (fasting), 5 mmol/L (4.2-6.1 mmol/L)
• Alanine aminotransferase [ALT], 1.8 μkat/L or 105 units/L (0.12-0.70 μkat/L or 7-41 units/L)
• Plasma iron, 50 μmol/L (7-25 μmol/L)
• Total iron-binding capacity, 55 μmol/L (45-73 μmol/L)
• Transferrin saturation with iron 82% (16-35%)
• Serum ferritin, 3200 μg/L, (29-248 μg/L, males)
X-rays of the joints showed loss of articular cartilage, narrowing of joint spaces, and diffuse demineralization.
In view of the above findings, it was decided to perform a liver biopsy. Histologic examination revealed moderate periportal fibrosis. Hemosiderin (aggregates of ferritin micelles) was visible as golden brown granules in both parenchymal and bile duct epithelial cells; with Prussian blue staining, iron was markedly visible in these cells. Quantitative measurement of iron in the biopsy material revealed a marked elevation of iron (8100 μg/g dry weight; normal: 300-1400 μg). The laboratory and other findings were consistent with a diagnosis of hereditary hemochromatosis.
Treatment
The treatment of hereditary hemochromatosis is relatively simple, consisting of withdrawing blood from the patient until the excess iron in the body is brought down to near normal values. This is accomplished initially by weekly phlebotomy of approximately 500 mL of blood (250 mg of iron). The efficacy of treatment is assessed by monthly monitoring of serum ferritin and transferrin saturation. Once these parameters have reached satisfactory levels, phlebotomy is required only once every 3 months.
Chelation therapy (eg, with deferoxamine) is rarely required and is much more expensive, as well as less effective than phlebotomy. Foods high in iron should be avoided.
Complications such as hepatic cirrhosis, diabetes mellitus, infertility, and cardiac problems must be treated appropriately. Screening of family members is recommended in order to give them early treatment if necessary. If the condition is recognized early (eg, before hepatic cirrhosis develops), the prognosis is excellent. Hepatocellular carcinoma may develop in patients with cirrhosis. The arthropathy of hemochromatosis does not respond well to treatment and may continue relentlessly despite normalization of body iron parameters.
Discussion
This Section of Case 10 was written by Drs Joe Varghese and Moly Jacob.
The key feature of hemochromatosis is an increase in total body iron sufficient to cause tissue damage. Total body iron ranges between 2.5 and 3.5 g in normal adults; in hemochromatosis, it usually exceeds 15 g. It is critical to diagnose the condition early to prevent complications such as cirrhosis and liver cancer. Hepatomegaly, skin pigmentation, diabetes mellitus, heart disease, arthropathy, and hypogonadism are usually found in full-blown cases. However, it is common to find patients with only one or two of the above-mentioned features. Therefore, to achieve an early diagnosis, a high index of suspicion is necessary. Elevated levels of transferrin saturation and serum ferritin are the most useful tests for early diagnosis.
In hereditary hemochromatosis, the absorption of iron from the small intestine is greatly increased. There are no physiological mechanisms to remove excess iron from the body. Free iron is toxic because of its ability to generate free radicals (Chapter 45). The accumulated iron damages organs such as the liver, pancreatic islets, and the heart. Melanin and iron accumulate in the skin, accounting for the slate-gray color often seen. The precise cause of melanin accumulation is not clear. The frequent co-existence of diabetes mellitus (due to pancreatic islet damage) and the skin pigmentation led to use of the term bronze diabetes for this condition.
Hereditary hemochromatosis is an autosomal recessive disorder. It is common in Europe (carrier frequency of approximately 1:10), particularly in Ireland and Scotland. Emigrants from these countries have contributed to dissemination of the affected gene around the world. Since 1976, it has been known that there is an association between HLA antigens and hereditary hemochromatosis. In 1996, Feder and colleagues isolated a gene, now known as HFE, located on chromosome 6 (6p21.3) in close proximity to the major histocompatibility complex (MHC) genes. The encoded product was found to be related to the MHC class 1 antigens. HFE has been found to exhibit three missense mutations in individuals with hereditary hemochromatosis. The most frequent mutation is one that changes a cysteinyl residue at position 282 to a tyrosyl residue (C282Y), resulting in conformational changes in the structure of the protein. The other mutation involves a change of a histidyl residue at position 63 to an aspartyl residue (H63D). The incidence of these two mutations varies in different ethnic groups. C282Y is less frequent in Italians than in Northern Europe. A third less frequent mutation (S65C) has also been found, but has not as yet been studied in much detail. A small group of individuals are compound heterozygotes (C282Y/H63D).
HFE is normally expressed on the surface of cells, bound to β2-microglobulin and TfR1. The C282Y mutation inhibits the formation of this complex, resulting in decreased cell surface expression of the HFE protein. It was initially thought that disruption of the HFE-TfR1 interaction in the intestinal crypt cell was responsible for increased duodenal iron absorption seen in hereditary hemochromatosis. However, it is now known that the primary role of HFE is in the regulation of hepatic hepcidin expression (described in Chapter 50). Mutations in the HFE gene are associated with low circulating hepcidin levels. Hepcidin binds to and triggers the internalization and degradation of ferroportin, which is the exporter of iron from enterocytes, macrophages, and other cells. Low levels of hepcidin therefore increase ferroportin expression on the basolateral membrane of enterocytes. Mutations in genes other than HFE (hepcidin, TfR2, HJV) have also been implicated in hereditary hemochromatosis, though these are comparatively rare (see Table 50-5). The proteins encoded by these genes play important roles in hepatic hepcidin expression. A low plasma hepcidin level is a characteristic finding in these conditions. A tentative scheme of the main events in causation of hereditary hemochromatosis is given in Figure 57–14.
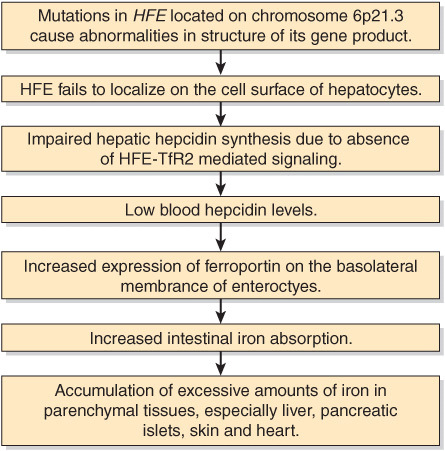
FIGURE 57–14 Tentative scheme of the main events in causation of hereditary hemochromatosis (OMIM 235200). The two principal mutations seen in HFE are CY282Y and H63D. Mutations in genes other than HFE also cause hemochromatosis.
Penetrance of hereditary hemochromatosis is low, and is lower in women than in men. Penetrance is the fraction of individuals with a genotype known to cause a disease that has signs and symptoms of the disease. Genetic testing has been evaluated but is not presently recommended, except in family members of patients with hereditary hemochromatosis. Tests for HFE mutations in individuals with elevated serum iron concentrations may be useful.
CASE 11: HYPOTHYROIDISM, PRIMARY
Before studying this case, the reader is advised to consult the material in Chapter 41 dealing with the thyroid gland.
Causation
Primary hypothyroidism is a state due to deficiency of the thyroid hormones, usually due to impaired function, damage to or surgical removal of the thyroid gland. Specific causes are discussed below.
History, Physical Examination, and Laboratory Tests
A 57-year-old woman visited her family physician complaining of chronic fatigue and sluggishness for a number of years; this was her first visit to her doctor for 5 years. On questioning, a history of constipation and feeling cold (cold intolerance) was elicited. She had two adult children; her last menstrual period had occurred some seven years previously. A sister had pernicious anemia and a maternal aunt had had “a thyroid problem.”
On examination, the patient was moderately obese. She answered questions slowly, with little change of expression, her voice sounded coarse, and her tongue appeared moderately swollen. Some puffiness around her cheeks was also evident. Palpation of the neck revealed that her thyroid gland was rather firm in consistency, and modestly enlarged. Her blood pressure was mildly elevated and her deep tendon reflexes were delayed. Some clinical findings in a case of hypothyroidism are summarized in Figure 57–15. On the basis of the history and clinical examination, her doctor suspected that the lady had hypothyroidism. She ordered various tests to investigate this possibility; the following results were these of relevance:
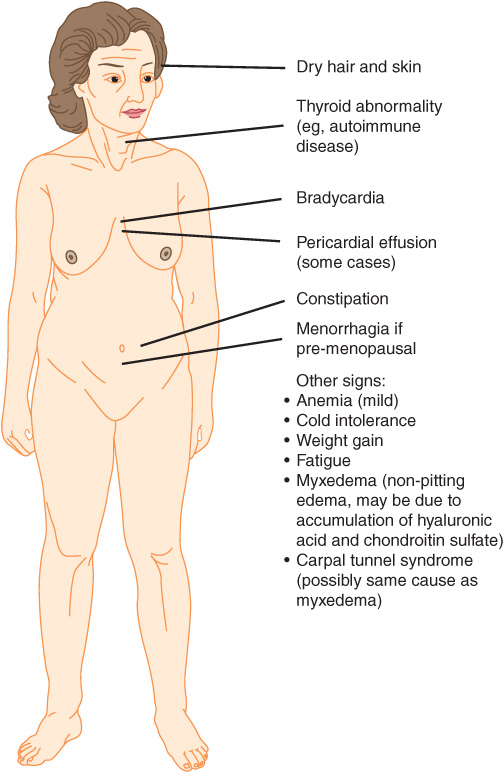
FIGURE 57–15 Some of the major signs of hypothyroidism.
• Thyroid stimulating hormone (TSH): 20 mIU/L (normal range, 0.34-4.25 mIU/L)
• Thyroxine (T4), free: 4.0 pmol/L (normal 10.3-21.9 pmol/L)
• Anti-thyroperoxidase (anti-TPO) antibodies: ++++ (normal trace)
• Cholesterol, total: 6.20 mmol/L (normal <5.17 mmol/L)
• Chest x-ray: Revealed a small pericardial effusion.
• ECG: Revealed bradycardia and low voltage complexes, but no evidence of ischemia or arrhythmias.
• Hemoglobin and RBC count: Results consistent with a mild normocytic anemia.
Treatment
The clinical history, physical examination, and lab results were all consistent with primary hypothyroidism. Accordingly, the patient was started on a low dose of thyroxine (T4). It is important to begin therapy with a small dose of T4, as larger doses can precipitate serious cardiac events, due to the changes in metabolism caused by administration of the hormone. The dosage of T4 is gradually increased at 6-8 week intervals, up to a maximum of approximately 125 μg. Assessment of progress is made by assays of TSH, which should eventually decline to normal range and be sustained there. Regular assessments are important. Once started, T4 therapy is generally continued for life.
Discussion
Primary hypothyroidism is a relatively prevalent condition, and is probably the most common endocrine problem (excluding diabetes mellitus) seen in clinical practice. The most frequent cause worldwide is deficient intake of iodine. In North America (as in the present case) and other developed countries, a major cause is Hashimoto disease, an autoimmune condition affecting the thyroid. Other causes include 131I ablation of the thyroid, surgical resection of the thyroid, and the use of drugs for treating hyperthyroidism. It is more common in females than in males. In this case, the diagnosis was relatively easy because of the classical history and clinical findings. However, it often has an insidious onset, developing gradually over years, and may not be considered. A case can be made for routine assay of TSH in everyone over 35 years of age, but there is as yet no consensus regarding this.
Secondary hypothyroidism is much less common, and is due to decreased secretion of TSH by various pathologic conditions affecting the pituitary gland. Pathology in the hypothalamus can cause tertiary hypothyroidism, due to decreased secretion of the hypothalamic thyroid-releasing hormone (TRH). Congenital hypothyroidism is usually due to various blocks in manufacture of the thyroid hormones and can result in cretinism if not diagnosed early and treated appropriately. All newborns in North America and many other countries are routinely screened for levels of TSH at birth.
Detection of increased levels of serum TSH is the most useful test for hypothyroidism. As levels of circulating thyroid hormones (T4, T3) drop due to thyroid destruction in Hashimoto disease, feedback inhibition on the pituitary declines and levels of TSH rise.
The presence of elevated TSH and decreased T4 is highly indicative of hypothyroidism. In Hashimoto disease, the thyroid gland becomes heavily infiltrated by lymphocytes and other inflammatory cells, which gradually destroy and replace much of the gland resulting in a progressive decrease of secretion of the thyroid hormones (see Chapter 41), causing the hypothyroid state. The lymphocytes include activated CD4+ T cells specific for various thyroid antigens. Various autoantibodies can be detected in serum from patients with Hashimoto disease; among these often measured at present are anti-thyroperoxidase (anti-TPO) antibodies, which serve as markers of Hashimoto disease. Very often there is a family history of the disease or of other autoimmune conditions, indicating a genetic contribution. In the present case, a sister of the patient’s mother had a family history of “thyroid disease” and a sister of the patient had pernicious anemia, another autoimmune condition.
It is important to consider hypothyroidism as a diagnosis, as early treatment can make a huge difference to a patient’s quality of life.
A simplified scheme of the causation of hypothyroidism is given in Figure 57–16.
FIGURE 57–16 Simplified scheme of the causation of primary hypothyroidism.
CASE 12: KWASHIORKOR, ONE TYPE OF PROTEIN-ENERGY MALNUTRITION (PEM)
Before studying this case, the reader is advised to consult the material in Chapters 43 and 44 on nutrition. Chapter 43 contains information on energy balance, kwashiorkor, marasmus, and essential amino acids.
Causation
Inadequate food intake, leading to deficiency of energy and also protein is the cause of both marasmus and kwashiorkor. Deficiency of antioxidant nutrients and the stress of infection precipitate kwashiorkor in undernourished children.
History and Physical Examination
A 2-year-old African girl was brought by her mother to the outpatient department of the local hospital. The mother had four children, the youngest of whom was 3 months of age and still being breast-fed. The father had broken his leg in an accident during the previous year and had not been able to work since. Family income was thus low, and they were not able to buy milk and meat on a regular basis. Their main subsistence food was a starchy gruel, high in carbohydrate and low in protein, and even that had been in short supply recently. The mother stated that the daughter had been eating poorly for the past several months, had intermittent diarrhea for that time, had recently developed a cough and fever, and had become very irritable, weak, and apathetic.
On examination, she was found to be both underweight for her height and small for her age. Her temperature was 40.5°C. Her midarm circumference was a little below normal. Her skin was flaky and her hair dry, brittle, and easily pluckable. The abdomen was distended and the liver moderately enlarged. Peripheral edema was evident. Rales were heard over the lower lobes of both lungs.
The doctor on duty made the diagnosis of kwashiorkor, diarrhea, pneumonia, and possible bacteremia.
Laboratory Findings
Samples of blood were taken for analysis. Results were subsequently reported as hemoglobin 6.0 g/dL (normal for a 2 year old: 11-14 g/dL), total serum protein 4.4 g/dL (normal 6.0-8.0 g/dL), and albumin 2.2 g/dL (normal 3.5-5.5 g/dL). Stool and blood samples were taken for culture; a gram-negative anaerobe was later reported in both. The neutrophil count was elevated (consistent with a bacterial pneumonia) and her lymphocyte count was markedly depressed. Chest x-rays revealed mottled opacities in the lower lobes of both lungs, consistent with bilateral acute bronchopneumonia.
Treatment
In many cases it is better not to treat children with mild to moderate kwashiorkor in the hospital, because this only increases the chance of infection. However, in view of the fever, weakness, drowsiness, and severe edema, this patient was admitted. She was immediately started on an appropriate antibiotic and intravenous saline-dextrose infusion. Sadly, her condition worsened and she died approximately 12 h after admission. Autopsy findings were compatible with kwashiorkor and also revealed severe fatty liver and bilateral bronchopneumonia.
Discussion
PEM is the most common nutritional disorder in many parts of the world. As many as one billion people suffer from various degrees of severity of PEM. It is due to inadequate dietary intake of protein and energy; kwashiorkor is commonly precipitated by the oxidative stress of an infection. PEM is almost always accompanied by deficiencies of other nutrients (eg, vitamins, minerals, etc). Children and the elderly are particularly susceptible, but it may occur at any age.
PEM may be defined as primary (due directly to deficiency of protein and energy intake), or secondary (due to increased nutrient needs, decreased absorption of nutrients, or increased loss of nutrients). This was a case of primary kwashiorkor.
Many of the features of primary PEM represent adaptations to the dietary deficiencies of energy and protein. For example, physical activity decreases in the face of deficient intake of nutrients. Stores of glycogen in muscle and liver are only capable of supplying energy for a short time (a day or two), so fat stores are mobilized to produce energy.
Eventually, when these are exhausted, protein is catabolized (mainly in muscles) to supply amino acids and energy. Thus, patients with PEM exhibit little activity, have diminished or no body stores of fat, and show muscle wasting, depending on the severity of the condition.
PEM has been classified as edematous (kwashiorkor) or nonedematous (marasmus). The precise cause of the edema in kwashiorkor is still under study. Hypoalbuminemia (due to deficient supply of amino acids to synthesize the protein) is likely one contributing factor (see Chapter 50), although this is not settled. Increased vascular permeability due to oxidative stress secondary to infection may also be important. Deficiency of the amino acid methionine, a precursor of cysteine, may also contribute. Cysteine is one of the three amino acids present in glutathione, the body’s major antioxidant. If the tissues levels of glutathione decline, this could result in free radicals damaging various molecules and tissues (see Chapter 45) and perhaps damaging cell membranes, increasing their permeability.
Marasmus is the predictable outcome of severe energy deficiency, and the affected child is less than 60% of expected weight for age. In kwashiorkor the child is 60-80% of expected weight for age, and is edematous. If the child is less than 60% of weight for age and edematous, this is marasmic kwashiorkor, the most severe and serious type of PEM. This patient exhibited primarily signs of kwashiorkor. The hallmarks of kwashiorkor are hypoalbuminemia, fragile skin (eg, poor wound healing, ulcers), easy hair pluckability, and edema (see Figure 57–17). Kwashiorkor is the word used by members of the Ga tribe in Ghana to describe “the sickness the older child gets when the next child is born.” It follows weaning from breast milk and exposure to a diet low in protein and high in carbohydrate. Fatty liver is often found in kwashiorkor because of depressed synthesis of apoliproteins in the liver, resulting in accumulation of triglycerides. The poor skin and hair seen in kwashiorkor are mainly due to protein deficiency. Hypoalbuminemia is a common feature. While deficiency of protein can cause hypoalbuminemia, chronic inflammation can also contribute by suppressing synthesis of albumin. Total iron-binding capacity and levels of transferrin are also depressed.
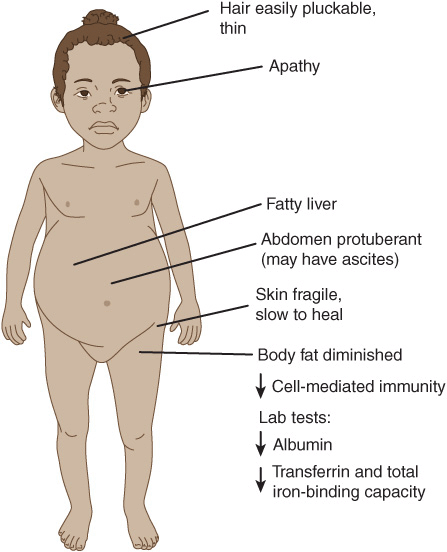
FIGURE 57–17 Some of the major signs of kwashiorkor.
Hormones may be important in the generation of PEM. The exposure to a relatively high intake of carbohydrate is thought by some to keep levels of insulin high and levels of epinephrine and cortisol low in kwashiorkor, as opposed to marasmus. The combination of low insulin and high cortisol greatly favors catabolism of muscle; thus, muscle wasting is greater in marasmus than in kwashiorkor. In addition, because of the lower levels of epinephrine, fat is not mobilized to the same extent in kwashiorkor. The immune system is impaired in PEM, particularly T-cell function. Individuals are thus very susceptible to infections (eg, causing diarrhea), and infections worsen the situation by placing a higher metabolic demand on the body (eg, through fever). Some differences between kwashiorkor and marasmus are summarized in Table 57-5.
TABLE 57–5 Some Differences between Kwashiorkor and Marasmus
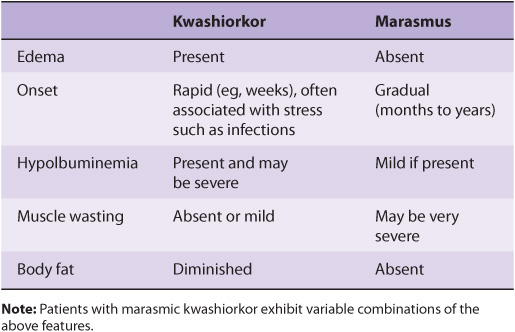
PEM is entirely preventable by a well-balanced diet containing adequate amounts of the major macronutrients, micronutrients, vitamins, and minerals.
Figure 57–18 summarizes some of the mechanisms involved in kwashiorkor.
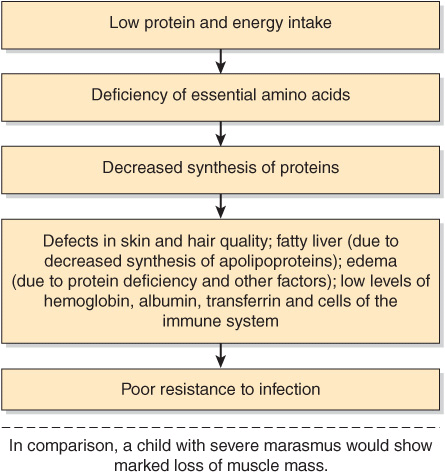
FIGURE 57–18 Summary of some of the factors involved in the causation of kwashiorkor.
CASE 13: MYOCARDIAL INFARCTION (MI)
Etiology
Lack of oxygen and various metabolites (due to blockage of blood flow in a coronary artery to an area of myocardium). Genetic and other factors predispose to this situation.
History and Physical Examination
A 46-year-old businessman was admitted to the emergency department of his local hospital complaining of severe retrosternal pain of 1-h duration. He had previously been admitted to the hospital once for treatment of a small MI, but despite this he continued to smoke heavily. He had been advised to take a mainly vegetarian diet, restrict his salt intake, join an exercise program, and prescribed an HMG-CoA reductase inhibitor (a statin) (his total cholesterol and LDL cholesterol had been elevated and HDL depressed), and a combination of a thiazide diuretic and an ACE (angiotensin-converting enzyme) inhibitor for moderate hypertension. He was also taking one aspirin (81 mg) per day. His blood pressure was 150/90 mmHg (before this incident it had been 140/80 mmHg; it was likely elevated because of stress), pulse was 60/min, and he was sweating profusely. There was no evidence of cardiac failure. His father had died at age 52 from a “heart attack,” and one of his two siblings had had an MI at age 49. Because of the admitting diagnosis of probable MI, he was given morphine to relieve his pain and apprehension and also for its coronary dilator effect, and immediately transferred to a cardiac care unit, where continuous electrocardiographic monitoring was started at once.
Laboratory Findings
The initial ECG showed S-T segment elevation and other changes in certain leads, indicative of an acute anterior transmural left ventricular infarction. Blood was taken initially and at regular intervals thereafter for measurement of troponin T; on admission the level was within normal limits, but had risen eightfold by 6 h after admission.
Some proteins and enzymes that have been used in the diagnosis of a myocardial infarction are indicated in Figure 57–19. The use of enzymes and proteins in the diagnosis of MI and other diseases is discussed in Chapter 7. Levels of total cholesterol and the LDL/HDL cholesterol ratio were within normal limits (<5.17 mmol/L and 4:1, respectively), and triacylglycerols were 1.50 mmol/L (normal <2.26 mmol/L).
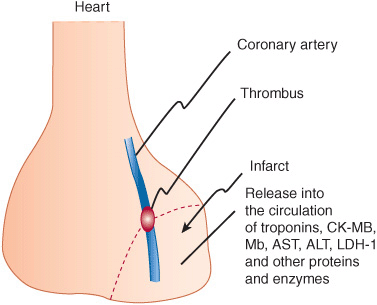
FIGURE 57–19 Diagrammatic representation of a coronary artery thrombus resulting in release of various proteins and enzymes into the circulation from an area of myocardial infarction (MI). Various proteins and enzymes are released from infarcted tissue at different rates, and they exhibit different half-lives in the circulation. At the present time, troponins are widely used to assist in the diagnosis of MI, but the other enzymes shown are still used to variable extents, and were more widely used in the past. (Mb, myoglobin; CK-MB, the MB isozyme of creatine kinase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; LDH-1, the heart muscle isozyme of lactate dehydrogenase.)
Treatment
The attending cardiologist, after reviewing all aspects of the case, decided to administer tissue plasminogen activator (t-PA) (see Chapter 51) intravenously because of the diagnosis of anterior transmural MI. Some 1.5 h had elapsed since the onset of symptoms. Chest pain began to disappear after 12 h, and the patient felt increasingly comfortable. He was discharged from hospital 7 days later under the care of his family doctor. He was instructed to continue his medication, to attend a cardiac rehabilitation program, and to stop smoking.
Discussion
An MI is generally caused by an occlusive thrombus lying in close proximity to an unstable atherosclerotic plaque that has often ruptured recently. The rupture of the plaque helps generate the thrombus. Usually the diagnosis can be made from the clinical history, the electrocardiographic results, and serial measurements of a cardiac biomarker, such as troponin T. Measurement of this protein has replaced that of CK-MB in many hospitals (see Chapter 7).
Major aims of treatment are to prevent death from cardiac arrhythmias by administration of appropriate drugs and to limit the size of the infarction. In this case, the decision was made to limit the size of the infarct by administration of t-PA, which can dissolve or limit the growth of the thrombus (see Chapter 51) if given up to 12 h of onset of symptoms, although preferably sooner. An alternative would have been percutaneous coronary intervention (PCI), consisting of percutaneous transluminal coronary angioplasty (PTCA), with or without insertion of a stent.
Atherosclerosis, coronary thrombosis, and MI are described here very briefly; a textbook of pathology should be consulted for detailed descriptions. Atherosclerosis consists of patchy plaques in the intima of medium and large arteries. Endothelial dysfunction is believed to play an important role in the genesis of atherosclerosis. Platelets and fibrin can deposit on the luminal aspect of an artery and monoclonally derived smooth muscle cells present in the medial layer of artery grow into the intimal lesion, attracted by growth factors released by macrophages and platelets (eg, platelet-derived growth factor). Plasma lipoproteins, glycosaminoglycans, collagen, and calcium subsequently accumulate in a lesion called a fatty streak. The presence of oxidized LDL in atherosclerotic lesions appears to be particularly important, in that it encourages recruitment of macrophages (inflammatory cells) and stimulates the release of growth factors. Inflammation is thus thought to be a key factor in atherosclerosis, as reflected by accumulation of macrophages and lymphocytes. The elevated plasma level of C-reactive protein (CRP) (Chapter 50) is also a reflection of chronic inflammation.
As the above processes progress, the fatty streak evolves into an intimal plaque Inflammation and hemorrhage into the plaque can occur, leading to rupture of its surface and exposure of its underlying constituents to the blood. Platelets will adhere to the exposed collagen, and a thrombus is initiated (Chapter 51).
Risk factors for atherosclerosis include age, family history, male sex, high levels of LDL and low levels of HDL, hypertension, diabetes mellitus, and smoking. This patient had elevated levels of total cholesterol and LDL and depressed levels of HDL before starting dietary and drug treatment.
If the thrombus in a coronary artery occludes approximately 90% of the vessel wall, blood flow through the affected vessel may cease (total ischemia) and the oxygen supply of the affected area of mycocardium will be rapidly compromised. The normal metabolism of the myocardium is aerobic, with most of its ATP being derived from oxidative phosphorylation. The anoxia secondary to total ischemia results in a switch to anaerobic glycolysis, which generates only about one-tenth of the ATP produced by oxidative phosphorylation. Not only does this switch in metabolism occur, but the flow of substrates into the myocardium via the blood and the removal of metabolic products from it are also greatly reduced. This accumulation of intracellular metabolites increases the intracellular oncotic pressure, resulting in cell swelling, affecting the permeability of the plasma membrane. Thus, the affected myocardium exhibits depletion of ATP, accumulation of lactic acid, development of severe acidosis, and marked reduction of contractile force. The precise metabolic changes that commit a cell to dying are being investigated in many laboratories; this is a very important area of research. Changes under study include depletion of ATP, activation of intracellular phospholipases (resulting in damage to cellular membranes), activation of proteases, and accumulation of intracellular Ca2+.
Some of the mechanisms involved in the causation of an acute MI are summarized in Figure 57–20.
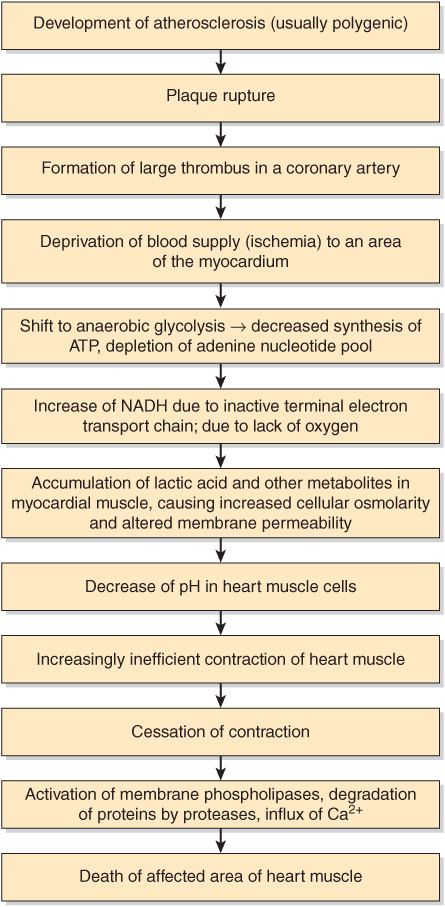
FIGURE 57–20 Summary of mechanisms involved in the causation of an acute myocardial infarction. The arrows do not in all cases imply a strict causal relationship.
CASE 14: OBESITY
Before studying this case history the reader is advised to consult the material on triacylglycerols and adipose tissue in Chapter 25 and the material on nutrition in Chapters 43 and 44.
Causation
Many factors contribute to obesity (genetic, environmental, cultural, etc). However, the central issue is that energy intake exceeds energy expenditure, resulting in the storage of triacylglycerols in adipose tissue.
History, Physical Examination, and Laboratory Findings
A 30-year-old woman visited her family doctor complaining of being markedly overweight. She was married, but had no children. She also complained that her periods were irregular. She gave no significant past history and was not on any drugs. She stated that she had always tended to being overweight, and that her mother and two sisters were also overweight. She had a sedentary job in an office and did not exercise regularly. In addition, she said that she had “a healthy appetite,” and both she and her husband (who was also overweight) often consumed a variety of fast foods. Many obese individuals deny that they eat excessively, and it is difficult to measure food consumption precisely in ordinary medical practice.
On examination, she was obviously overweight (200 lb, 91 kg) for her size (5’4”, 163 cm). Her body mass index (BMI = weight kg/height m2) was calculated from a table and found to be ~34. A BMI of 25-29.9 kg/m2 indicates overweight and a BMI of >30 kg/m2 indicates obesity. Other clinical indicators of obesity include waist-hip ratio and skin fold thickness. If fat accumulates around the abdomen this confers an apple shape, whereas fat accumulating around the buttocks confers a pear shape. The former may be more serious, as abdominal (visceral) fat upon lipolysis may release fatty acids into the portal vein, leading to fat deposition in the liver and muscles. More precise tools for measuring obesity are also available (eg, bioelectrical impedance analysis and underwater weighing). Her blood pressure was 140/95 and her total cholesterol was 6.1 mmol/L (borderline high). Urinalysis was negative. Her fasting blood glucose was 6.6 mmol/L (borderline high).
Treatment
Her physician advised her that she was obese, but not morbidly obese ![]() . The laboratory results indicated that her levels of cholesterol and blood glucose were borderline high, as was her blood pressure. He told her that further elevation of these values would predispose her to heart disease, diabetes mellitus, hypertension, and the metabolic syndrome. The latter is characterized by excess abdominal fat, high blood glucose (resistance to insulin), abnormal blood lipids (increased LDL and decreased HDL), and high blood pressure. He also pointed out a number of other complications to which obesity predisposed her (eg, reproductive problems such as irregular periods, gallbladder disease, deep vein thrombosis, sleep apnea, etc). He indicated to the patient that treatment of obesity was not easy, and that she would have to undergo a permanent change in lifestyle if she wished therapy to be successful and weight loss to be maintained. The major approaches to treatment of obesity were summarized: (1) diet, (2) exercise, (3) behavioral therapy, (4) drugs (eg, to suppress appetite centrally, or acting as an inhibitor of lipase activity in the gut, thus reducing absorption of fatty acids), and (5) surgery (eg, laparoscopic adjustable gastric banding and other procedures) in very severe cases.
. The laboratory results indicated that her levels of cholesterol and blood glucose were borderline high, as was her blood pressure. He told her that further elevation of these values would predispose her to heart disease, diabetes mellitus, hypertension, and the metabolic syndrome. The latter is characterized by excess abdominal fat, high blood glucose (resistance to insulin), abnormal blood lipids (increased LDL and decreased HDL), and high blood pressure. He also pointed out a number of other complications to which obesity predisposed her (eg, reproductive problems such as irregular periods, gallbladder disease, deep vein thrombosis, sleep apnea, etc). He indicated to the patient that treatment of obesity was not easy, and that she would have to undergo a permanent change in lifestyle if she wished therapy to be successful and weight loss to be maintained. The major approaches to treatment of obesity were summarized: (1) diet, (2) exercise, (3) behavioral therapy, (4) drugs (eg, to suppress appetite centrally, or acting as an inhibitor of lipase activity in the gut, thus reducing absorption of fatty acids), and (5) surgery (eg, laparoscopic adjustable gastric banding and other procedures) in very severe cases.
He stated that in her case he believed satisfactory progress could be made, at least initially, through use of the first two lines of therapy. He told the patient that he seldom recommended drugs for treatment of obesity, and that surgery was generally restricted to people with morbid obesity (BMI >40) who had not responded to other approaches. He also encouraged her to get her husband involved because of his weight problem, and because it would be mutually beneficial if he also were on a weight reduction program.
Regarding diet, her doctor (who had a special interest in implementing healthful nutrition in his practice) discussed general features of a suitable diet and also the advantages of regular daily exercise. Specific dietary recommendations that he made are listed in Table 57-6.
TABLE 57–6 Summary of Dietary Advice Regarding Weight Loss


She subsequently joined a weight loss organization and found that it stressed changes in behavior (eg, developing sensible eating habits, rewards for good results, group counseling) and also provided support and encouragement from the other members. The patient was very keen to lose weight, and over the next year she closely followed the various advices given. She lost 34 lbs over that period of time. She felt much better and also stated that her periods had regularized and she was hoping to get pregnant. In addition, her blood pressure and her levels of total cholesterol and blood glucose were down to normal values. She was absolutely determined to continue on the weight loss program, as was her husband. Her sole regret was that she had not lost even more weight. Many patients under treatment for obesity eventually regain lost weight, for a variety of reasons.
Discussion
Obesity is a very prevalent condition, which is on the increase. Almost one third of adults in the Unites States are obese, and, alarmingly, more and more children are obese. Some talk of an epidemic of obesity in Western society. Dissecting all the factors contributing to this is complex, but increased energy intake and decreased energy expenditure play important roles. The increased intake of fast foods and high calorie soft drinks and TV watching by “couch potatoes” are important contributors.
While the dangers of obesity have been widely circulated, it is of interest that some obese individuals feel that they are being unfairly targeted by health professionals and that the dangers of obesity have been overemphasized. It can be debated that it is better to be somewhat obese and fit, than to be of normal weight and unfit. In addition, certain individuals claim that they enjoy being obese.
While obesity itself is relatively easy to recognize and define (eg, we can somewhat arbitrarily use a ![]() ), pinning down the specific factors that contribute to individual cases is not that easy.
), pinning down the specific factors that contribute to individual cases is not that easy.
In the present case, a number of contributory factors are apparent. For example, she led a sedentary lifestyle, she frequently ate fast foods, she did not exercise regularly, etc. However, what was her precise caloric intake? What was her precise energy expenditure? Were the various mechanisms controlling appetite (see Figure 57–21) working properly? What role if any did genetic factors play in her obesity (she gave a family history of obesity)? These factors are not easy to quantify for a physician in her/his office.
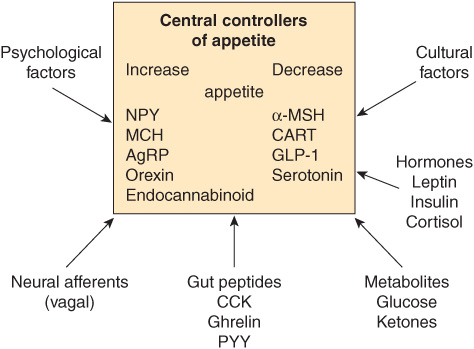
FIGURE 57–21 Factors that regulate appetite through effects on central neural circuits. Factors that increase or decrease appetite are listed. (NPY, neuropeptide Y; MCH, melanin-concentrating hormone; Ag RP, Agouti-related peptide; MSH, melanocyte-stimulating hormone; CART, cocaine- and amphetamine-related transcript; GLP-1, glucagon-related peptide-1, CCK, cholecystokinin; PYY, peptide YY.) The reader is advised to consult a textbook of Physiology for descriptions of the actions of these various molecules. (Reproduced, with permission, from Harrison’s Principles of Internal Medicine, 17th ed, Fauci AS et al [editors], McGraw-Hill, 2008.)
Because of its medical importance, much research is going on presently in the field of obesity. This spans basic research on the adipocyte and signaling mechanisms to epidemiologic studies on various populations and their susceptibility to obesity. Here we shall only touch briefly on three areas: (1) regulation of appetite and food intake; (2) some genetic aspects; and (3) some aspects of energy expenditure.
Figure 57–21 summarizes a body of knowledge concerning the regulation of appetite. The hypothalamus plays a key role in the central regulation of appetite. Factors that both increase and suppress appetite are shown. Psychologic, neural, and cultural factors all play a role. A variety of peptides, affecting specific areas of the hypothalamus, are indicated. In addition, levels of circulating metabolites (eg, glucose) and of hormones affect the hypothalamic centers. Regarding hormones, for example, individuals with Cushing syndrome (high levels of cortisol or related hormones) are obese and display a characteristic distribution of body fat. Particular interest has focused on leptin, a polypeptide released from adipocytes that acts mainly on the hypothalamus. Elevated levels of leptin decrease food intake and also increase energy expenditure. Leptin was discovered through studies of genetically obese mice (ob/ob). In humans, leptin is the product of the OB gene. The levels of leptin are elevated in most obese humans, suggesting that in some manner they may be resistant to its action.
Genetic influences on obesity have been recognized. Identical twins tend to have similar body weights. Occasional cases of mutations in the genes encoding leptin and the leptin receptor have been identified. Cases of obese individuals having mutations in the gene encoding proopiomelanortin (POMC), which is processed to form α-melanocyte hormone (α-MSH), a powerful appetite suppressant, have been described. In addition, mutations in the gene encoding the type-4 receptor for α-MSH and in the genes encoding two proteolytic enzymes involved in the conversion of POMC to α-MSH have been reported.
Children with the Prader-Willi syndrome (due to deletion of a part of chromosome 15; affected individuals overeat, among other signs) and with the Laurence-Moon-Biedl syndrome (an autosomal recessive genetic disorder) are characteristically obese. It should be stressed that the great majority of obese individuals do not apparently have mutations in the genes referred to above. However, it is likely that multiple subtle genetic factors (eg, single nucleotide polymorphisms, SNPs) influence obesity.
Regarding energy expenditure, like food intake, this is difficult to measure precisely except in research facilities. A sophisticated method for measuring energy expenditure using doubly labeled water is described in Chapter 44, as are the concepts of basal metabolic rate (BMR) and other factors involved in daily energy expenditure. It appears that most obese individuals have higher energy expenditure than people of normal weight, because their lean body mass is increased. Another important issue is whether obese individuals actually eat more than non-obese individuals. It appears likely that they do although many deny this. In the present case, it seems likely from her history that the patient did overeat.
An issue that has been the subject of debate is the possible role of variations in diet-induced thermogenesis (see Chapter 25) in contributing to obesity. Brown adipose tissue contains a mitochondrial protein known as thermogenin (uncoupling protein-1) that dissipates energy as heat. While brown adipose tissue is not a prominent component of adults (unlike newborns), it does occur. Also it appears to be diminished in amount in at least certain obese individuals, which could mean that they dissipate less energy as heat than non-obese individuals and have more available for other purposes. Two other uncoupling proteins are known to exist in human tissues, although their overall contribution to diet-induced thermogenesis is not clearly established. Thus, many factors can contribute to obesity and it is not easy to assess their contribution in most cases seen in clinical practice. At the present time, it is reasonable to treat obesity primarily by reducing intake of food, eating a healthful diet, increasing physical activity and offering appropriate support and encouragement. One way in which ethanol may disturb the functions of these proteins is by replacing water molecules at various critical sites.
Figure 57–22 summarizes some major factors involved in the causation of obesity.
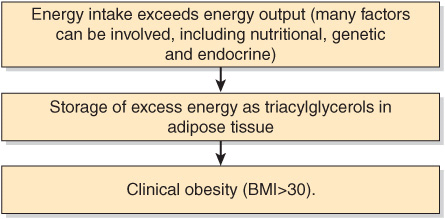
FIGURE 57–22 Simplified scheme of the causation of obesity.
CASE 15: OSTEOPOROSIS, PRIMARY (POSTMENOPAUSAL)
Before studying this case history the reader is advised to peruse the material in Chapter 48 on bone and the material in Chapter 44 on vitamin D.
Causation
Osteoporosis is loss of bone mass with preservation of the normal ratio of organic matrix (mostly proteins) to mineral. A variety of factors (endocrine, nutritional, lack of physical activity, etc) contribute to its development. In the postmenopausal type of osteoporosis dealt with here, estrogen deficiency is the major factor.
History, Physical Examination, and Investigations
A 64-year-old woman presented at emergency department, having tripped in her garden and apparently fallen rather gently onto her right forearm. Nevertheless, she suspected that she had fractured a bone in her arm, because of the pain and swelling just above her right wrist joint. X-rays showed a fracture of the distal end of the radius, with displacement. The radius also showed moderate reduction of radiodensity, suggestive of osteoporosis. The fracture was reduced, an appropriate cast was applied and she was asked to report to her family physician 2 weeks later. The emergency department physician gave her a note to give to her own physician, which mentioned that, because of the mildness of her fall, the resultant fracture, and the reduced radiodensity of her radius, he suspected that she might have osteoporosis. The patient attended her family doctor 2 weeks later. She had four adult children. Her last menstrual period had been some 5 years previously, and she had only attended her physician very irregularly over the years for the occasional minor ailment. She was not on any medication, did not take any vitamin or mineral supplements, and had never taken hormonal treatment for menopause. She ate very few fruits and vegetables, and overall she consumed a high carbohydrate diet along with liberal amounts of fried foods. She smoked one pack of cigarettes a day and also had several drinks of vodka each evening. In addition, she rarely exercised. She complained of chronic lower backache, but otherwise gave no significant history. In view of the fracture and the suggestion of osteoporosis, her family doctor ordered dual-energy x-ray absorptiometry (DEXA) of the lumbar spine and hip areas to assess bone density. He also ordered x-rays of the lower spine because of the history of lower back pain. In addition, he ordered determinations of serum Ca, P, alkaline phosphatase, 25-hydroxyvitamin D, parathyroid hormone, and complete urinalysis (including 24-h calcium) to check for any other bone disease (eg, due to vitamin D deficiency, hyperparathyroidism, or multiple myeloma). The results of her DEXA scan showed a marked reduction (over 3 standard deviations; over 2.5 is diagnostic for osteoporosis) from the average value in 25-year-old women of her race, compatible with severe osteoporosis. The X-rays of her lower spine showed decreased radiodensity, but no fractures. Results of her blood and urine tests were within normal limits, suggesting that she had no other serious bone disorder.
When bone resorption occurs, there is increased production of collagen cross-link products. These include N-telopeptide, deoxypyridinoline and C-telopeptide. Also, in some cases of osteoporosis, increased bone formation occurs; bone alkaline phosphatase and osteocalcin are markers of this. These various markers are not in themselves diagnostic of osteoporosis, but can be measured in serum or urine as indicators of response to therapy. For example, a 30% decrease of these markers would suggest successful therapy. They were not measured in this case, and in fact are not measured in many clinical laboratories.
Treatment
The history, DEXA scan, and the results of the other tests were consistent with a diagnosis of relatively severe osteoporosis. She was advised to start on an exercise program immediately at a gym, initially under the supervision of a personal trainer. She was also referred to a dietitian to change her dietary habits; recommendations included eating regular, daily portions of fresh fruits and vegetables, and consuming a more balanced diet (eg, reducing the starchy and fried foods). It was recommended that she stop smoking and cut down on her consumption of alcohol, as both of these may contribute to osteoporosis. She was also started on appropriate doses of calcium citrate and vitamin D. In addition to the above, her physician recommended that she start on a bisphosphonate (eg, alendronate or risedronate), giving detailed instructions as to how to take the drug. These two drugs, N-containing bispho-sphosphonates, are taken up by osteoclasts and inhibit formation of farnesyl diphosphate. This in turn inhibits prenylation of certain proteins (see Figure 26–2) and their attachment to the plasma membrane, negatively affecting osteoclast activity and thus inhibiting bone resorption. Other drugs that may be used in the treatment of osteoporosis in selected cases include salmon calcitonin, estrogen, selective estrogen modulators (eg, raloxifene), and parathyroid hormone. Estrogen was formerly widely prescribed early in menopause to reduce osteoporosis, and appeared to be relatively effective. However, the Women’s Health Initiative Study indicated that the risks of estrogen therapy outweighed the benefits in this situation.
The patient was followed up over the next few years. She lost considerable weight and maintained her exercise program. In general, she felt much more healthy than she had done in the previous 20 years. Her fracture healed satisfactorily, no further loss of bone mass occurred, but normal bone mass was not restored. She was advised on how to take precautions to avoid falls and it was also recommended that she wear hip pads.
Discussion
Osteoporosis can be defined as reduction of bone mass or density. Often it is first detected after a fracture, as the loss of bone tissue predisposes to fractures. The World Health Organization (WHO) has suggested that osteoporosis exists when bone density falls 2.5 standard deviations or more below the mean for young healthy adults of the same race and gender. In the United States, some 8 million women and 2 million men have osteoporosis, and many others are at risk of developing it.
Two terms related to osteoporosis are osteopenia and osteomalacia. Osteopenia is decreased bone mass, embracing both osteoporosis and osteomalacia. In osteoporosis, bone mass decreases due to decreased bone formation and increased resorption, but a normal ratio of bone mineral (hydroxyapatite) to bone matrix (mostly collagen type 1) is preserved. Osteomalacia is also an example of osteopenia, but in it there is decreased mineralization; its most common cause is deficiency of vitamin D.
Primary osteoporosis can be divided into three types: idiopathic (uncommon, and occurs in children and young adults), postmenopausal, and involutional (elderly) types. This case is an example of postmenopausal osteoporosis, and decline in estrogen levels is a major factor in its causation. This type can also occur in males due to a decline of serum testosterone, which acts to increase osteoclastic activity. Involutional osteoporosis occurs in older individuals and is due to the decline of the number of osteoblasts with age. Postmenopausal and involutional osteoporosis may coexist.
Secondary osteoporosis is relatively uncommon, and may be due to a variety of conditions (eg, chronic renal disease, various drugs [especially corticosteroids], a number of endocrine disorders, malabsorption syndrome, etc).
Many factors are involved in the causation of osteoporosis (see Figure 57–23). An in-depth understanding of them requires knowledge of bone modeling, of a variety of cytokines, of the actions of a number of hormones, and of various nutritional and genetic factors, among other considerations. (Cytokines are a heterogeneous group of proteins released by various cells and which have autocrine or paracrine effects.) Here we only indicate the complexity of a complete understanding of osteoporosis by briefly mentioning the main players. The central point is that osteoporosis occurs when bone resorption (osteoclastic [OC]) activity) exceeds bone formation (osteoblastic [OB] activity). In regards to post-menopausal osteoporosis in particular, one important consideration is that estrogen loss appears to increase the secretion of a number of cytokines that lead to recruitment of osteoclasts. Also, estrogen loss diminishes the secretion of certain other cytokines that promote osteoblastic activity. The overall effect is thus an imbalance between osteoclasts and osteoblasts, in favor of the former.
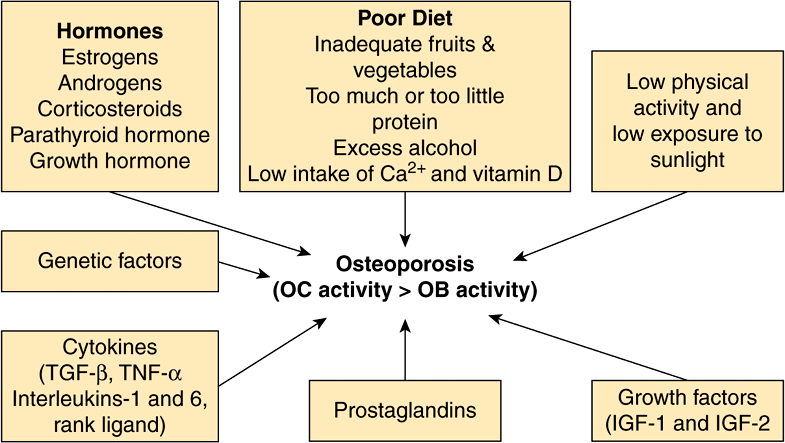
FIGURE 57–23 Simplified scheme of various factors involved in the causation of osteoporosis. (OC, osteoclastic, OB, osteoblastic, IGF-1 and IGF-2, insulin-like growth factors 1 and 2; TGF-β, transforming growth factor β; TNF-α, tumor necrosis factor α.) The hormones listed have a variety of effects on bone. IGF-1 and IGF-2 have anabolic effects on bone, but may also stimulate turnover of bone. RANK-ligand is a cytokine that is involved in communication between osteoblasts and osteoclasts; when it interacts with osteoclasts, it increases their activity. The other cytokines are made by osteoblasts. Their synthesis is increased or decreased by estrogen deficiency, with the overall effect of extending the life span of osteoclasts (by inhibiting apoptosis).
Figure 57–24 is a simplified schematic representation of the causation of osteoporosis.

FIGURE 57–24 Simplified scheme of some major factors in the causation of osteoporosis.
CASE 16: XERODERMA PIGMENTOSUM (XP) (PIGMENTED DRY SKIN)
The reader should consult the material in Chapter 35 on DNA repair and on XP before reading this case.
Causation
Genetic (a mutation in a gene directing synthesis of a DNA repair enzyme in the nucleotide excision repair pathway) and physical (exposure to ultraviolet [UV] irradiation) (Figure 57–25).

FIGURE 57–25 Summary of mechanisms involved in the causation of xeroderma pigmentosum (OMIM 278730 and other entries).
History and Physical Examination
An 8-year-old boy, an only child, presented at a dermatology clinic with a skin tumor on his right cheek. He had always avoided exposure to sunlight because it made his skin blister. His skin had scattered areas of hyperpigmentation and other areas where it looked mildly atrophied. There was no family history of a similar disorder. Because of the presence of a skin tumor at such a young age, the history of avoidance of sunlight, and the other milder skin lesions, the dermatologist made a provisional diagnosis of XP.
Laboratory Findings
Histologic examination of the excised tumor showed that it was a squamous cell carcinoma (a common type of tumor skin cancer in older people, but very unusual in a boy of this age). A small piece of skin was removed for preparing fibroblasts to be grown in tissue culture. A research laboratory in the hospital specialized in radiation biology and was set up to measure the amount of thymine dimers (see below) formed following exposure to UV light. The patient’s fibroblasts and control fibroblasts were exposed to UV light, and cell samples were taken at 8-h intervals for a total of 32 hours postirradiation. Extracts of DNA were prepared and the numbers of dimers remaining at each time point indicated were determined. Whereas only 24% of the dimers formed persisted in DNA extracted from the normal cells at 32 h, approximately 95% were found in the extract from the patient’s cells at 32 h. This showed that the UV-induced lesions had not been repaired, and thus confirmed the diagnosis of XP.
Discussion
XP is a rare autosomal recessive condition in which the mechanisms for repair of DNA subsequent to damage by UV irradiation are defective. This arises because of mutations in the genes encoding components of the nucleotide excision pathway of DNA repair (nucleotide excision repair, NER; see Chapter 35). The major damage inflicted on DNA by UV irradiation is the formation of thymine (pyrimidine) dimers (also known as cyclobutane pyrimidine dimers), where covalent bonds are formed between carbon atoms 5 and 5 and 6 and 6 of adjacent intrachain thymine residues, resulting in the dimers. Other types of damage can also occur.
NER has two subpathways: global genome repair and transcription-coupled repair. The former scans the whole genome and removes damaged areas rapidly. The second is linked to transcription, operates slowly, and removes damage from the transcribed strand of DNA.
Detailed analyses of NER involved in the removal of thymine dimers have been performed. The pathway is highly conserved across species, indicating its importance. In general, the pathway involves four major steps, all involving various enzymes: (1) recognition of damaged DNA; (2) excision of damaged region; (3) filling in of the gap by DNA polymerase; and (4) ligation of the filled in area. In E coli, an endonucleolytic cleavage, catalyzed by a specific endonuclease (also called an excinuclease), occurs on either side of the damage, releasing a 12- to 13-base-pair oligonucleotide. The polymerization step involves DNA polymerase and the final step is sealing by DNA ligase.
The NER pathway operates in humans, and its details are still being elucidated. It appears to be generally similar to the pathway in E coli. The most noticeable difference is that a much larger oligonucleotide (about 30 bases) is excised in humans. The products of at least seven genes (these encoding XPA through XPG) have been implicated in NER in humans, and all have been cloned. Mutations in any one of these genes cause XP. In the boy whose situation is discussed here, the specific gene involved was not determined.
Because the responsible genes have been cloned, prenatal diagnosis of XP is now possible using appropriate probes. The NER pathway is also involved in processes other than DNA repair, such as recombination, replication, and transcription. The involvement of the seven genes mentioned above in DNA repair was originally shown in the following manner. It was observed that when cultured cells from individuals with XP were cocultured with cells from other individuals under conditions in which cell fusion occurred, the defect in DNA repair could sometimes be corrected. This indicated that one set of cells was providing a normal gene product to the other, thus correcting the defect. In this manner, at least seven complementation groups, corresponding to the seven genes and their products mentioned above, have been recognized.
If UV damage is not repaired, mutations in DNA will result, chromosomal abnormalities may occur and cancer may ensue. Patients with XP often suffer from a variety of skin cancers from an early age.
As described in Chapter 35, there are other pathways of DNA repair operative in humans. They are all important in preserving the integrity of DNA, and abnormalities of some of them have been associated with cancer (eg, mismatch repair).
The parents of the boy in the present case were told that he would have to be watched closely throughout life for the development of new skin cancers. In addition, he was advised to avoid sunlight and to use an appropriate sunscreen ointment. Although XP is a rare condition, the existence of a variety of mechanisms for repairing DNA following exposure to different types of irradiation and to chemical damage is of great protective importance. Without their existence, life on this planet would be fraught with even more danger that it presently is! For instance, it has been estimated that patients with XP have a 1000-fold greater chance of developing skin cancer than do normal individuals.
Figure 57–25 summarizes the mechanisms involved in the causation of XP.
EPILOG
Remarkable progress has been made in biochemistry and in related fields such as genetics and cell biology. Many of the discoveries in these disciplines have had great impact on medicine and related life sciences. Crucial insights into the natures of many diseases have emerged from studies of their molecular bases. New drugs and other treatments are constantly being developed based on such discoveries. However, it is obvious that there are still many major challenges facing medical science. Table 57-7 summarizes some of them. The authors of this text and other biochemists believe that the application of biochemical, genetic, and allied approaches to these problems and to others not listed will pay rich dividends from which people all across the globe will benefit. Hopefully some of the readers of this text will contribute to such endeavors.
TABLE 57–7 Some Major Challenges Facing Medicine and Allied Health Sciences1


REFERENCES
Aiuti A, et al: Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 2009;360:447.
Beers MH, Porter RS, Jones TV, et al (editors): The Merck Manual of Diagnosis and Therapy. 18th ed. Merck Research Laboratories, 2006. (This book is available free online at http://merck.com/mmpe/index.html and contains coverage of many of the conditions discussed here).
Brunicardi FC, Anderson D, Billiar T, et al: Schwartz’s Principles of Surgery. 9th ed. McGraw-Hill, 2009. (Chapter 29 contains a discussion of colorectal cancer).
Brunton LL, Blumenthal D, Buxton I, et al: Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. McGraw-Hill, 2005 (Chapter 22 contains a discussion of acute ethanol intoxication).
Burtis CA, Ashwood ER, Bruns DE (editors): Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 4th ed. Elsevier Saunders, 2006. (Chapters 23 [Tumor Markers] and 49 [Mineral and Bone Metabolism] are of particular interest to the contents of this Chapter.)
Fauci AS, Braunwald E, Kasper DL, et al (editors): Harrison’s Principles of Internal Medicine. 17th ed. McGraw-Hill, 2008. (Contains comprehensive descriptions of many of the conditions described here).
Freedman DH: How to Fix the Obesity Crisis. Sci American 2011; 304 (No. 2):40.
Kohn DB, Candotti F: Gene therapy fulfilling its promise. N Engl J Med 2009;360:518. (Discusses aspects of the successful treatment of ADA deficiency.)
Neogi T: Gout. N Engl J Med 2011;364 (No. 5):443.
Riordan JR: CFTR function and prospects for therapy. Annu Rev Biochem 2008;77:70.
Scriver CR, Beaudet AL, Valle D, et al (editors): The Metabolic and Molecular Bases of odf Inherited Disease. 8th ed. McGraw-Hill, 2001. (The online version of this text contains comprehensive up-dated descriptions of many of the conditions discussed in this Chapter).
Shils ME, Shile M, Ross AC, et al (editors): Modern Nutrition in Health and Disease. 10th ed. Lippincott Williams & Wilkins, 2006. (Contains comprehensive coverage of nutritional topics, including PEM [Chapter 57] and obesity [Chapters 63 & 64]).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


