Bioavailability – physicochemical and dosage form factors
Marianne Ashford
Chapter contents
Physicochemical factors influencing bioavailability
Dosage form factors influencing bioavailability
Influence of the type of dosage form
Key points
• Lipid solubility and the drug dissociation affect drug absorption.
• Drugs need to dissolve before they are absorbed.
Introduction
As discussed in Chapter 19, the rate and extent of drug absorption are influenced by the physiological factors associated with the structure and function of the gastrointestinal tract. This chapter discusses the physicochemical properties of the drug and dosage form factors that influence bioavailability. For a drug to be absorbed, it needs to be in solution and to be able to pass across the membrane. In the case of orally administered drugs, this is the gastrointestinal epithelium. The physicochemical properties of the drug that will influence its passage into solution and transfer across membranes include its dissolution rate, pKa, lipid solubility, chemical stability and complexation potential.
Physicochemical factors influencing bioavailability
Dissolution and solubility
Solid drugs need to dissolve before they can be absorbed. The dissolution of drugs can be described by the Noyes–Whitney equation (Eqn 20.1). This equation, first proposed in 1897, describes the rate of diffusion of solute through boundary layers surrounding a dissolving spherical particle. When the dissolution process is diffusion controlled and involves no chemical reaction then this also equates to the rate of dissolution:
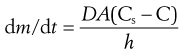 (20.1)
(20.1)
where dm/dt is the rate of dissolution of the drug particles, D is the diffusion coefficient of the drug in solution in the gastrointestinal fluids, A is the effective surface area of the drug particles in contact with the gastrointestinal fluids, h is the thickness of the diffusion layer around each drug particle, Cs is the saturation solubility of the drug in solution in the diffusion layer and C is the concentration of the drug in the gastrointestinal fluids.
More detail regarding Noyes–Whitney equation and its limitations in describing the dissolution of drug particles are outlined in Chapter 2. Despite these limitations, the equation serves to illustrate and explain how various physicochemical and physiological factors can influence the rate of dissolution in the gastrointestinal tract. These are summarized in Table 20.1 and are discussed in more detail in the next section.
Table 20.1
| Factor | Physicochemical parameter | Physiological parameter |
| Effective surface area of drug | Particle size, wettability | Surfactants in gastric juice and bile. pH, buffer capacity, bile, food components |
| Solubility in diffusion layer | Hydrophilicity, crystal structure | |
| Amount of drug already dissolved | Solubilization | Permeability, transit |
| Diffusivity of drug | Molecular size | Viscosity of luminal contents |
| Boundary layer thickness | Motility patterns and flow rate | |
| Volume of solvent available | Gastrointestinal secretions, co-administered fluids |
Figure 20.1 illustrates the dissolution of a spherical drug particle in the gastrointestinal fluids.
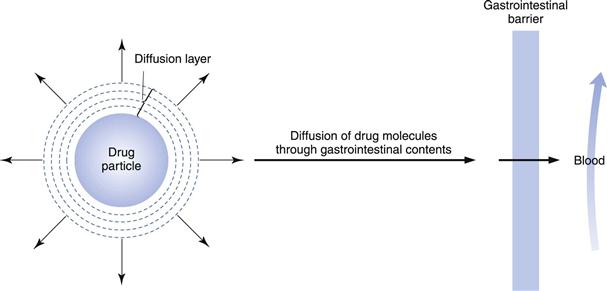
Fig. 20.1 Schematic representation of the dissolution of a drug particle in the gastrointestinal fluids.
Physiological factors affecting the dissolution rate of drugs
The environment of the gastrointestinal tract can affect the parameters of the Noyes–Whitney equation (Eqn 20.1) and hence the dissolution rate of a drug. For instance, the diffusion coefficient, D, of the drug in the gastrointestinal fluids may be decreased by the presence of substances that increase the viscosity of the fluids. Hence the presence of food in the gastrointestinal tract may cause a decrease in drug dissolution rate by reducing the rate of diffusion of the drug molecules away from the diffusion layer surrounding each undissolved drug particle. Surfactants in gastric juice and bile salts will affect both the wettability of the drug, and hence its effective surface area, A, exposed to gastrointestinal fluids, and the solubility of the drug in the gastrointestinal fluids via micellization. The thickness of the diffusion layer, h, will be influenced by the degree of agitation experienced by each drug particle in the gastrointestinal tract. Hence, an increase in gastric and/or intestinal motility may increase the dissolution rate of a sparingly soluble drug by decreasing the thickness of the diffusion layer around each drug particle.
The concentration of drug in solution in the bulk of the gastrointestinal fluids, C, will be influenced by such factors as the rate of removal of dissolved drug by absorption through the gastrointestinal-blood barrier and by the volume of fluid available for dissolution, which in turn will be dependent on the location of the drug in the gastrointestinal tract and the timing with respect to meal intake. In the stomach, the volume of fluid will be influenced by the intake of fluid in the diet. According to the Noyes–Whitney equation, a low value of C will favour more rapid dissolution of the drug by virtue of increasing the value of the term (Cs − C). In the case of drugs whose absorption is dissolution-rate limited, the value of C is normally kept very low by absorption of the drug. Hence dissolution occurs under sink conditions; that is, under conditions such that the value of (Cs − C) approximates to Cs. Thus for the dissolution of a drug in the gastrointestinal tract under sink conditions, the Noyes–Whitney equation can be expressed as:
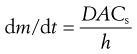 (20.2)
(20.2)
Drug factors affecting dissolution rate
Drug factors that can influence the dissolution rate are the particle size, the wettability, the solubility and the form of the drug (whether a salt or a free form).
Surface area and particle size.
According to Equation 20.1, an increase in the total surface area of drug in contact with the gastrointestinal fluids will cause an increase in dissolution rate. Provided that each particle of drug is intimately wetted by the gastrointestinal fluids, the effective surface area exhibited by the drug will be inversely related to the particle size of the drug. Hence the smaller the particle size, the greater the effective surface area exhibited by a given mass of drug and the higher the dissolution rate. Particle size reduction is thus likely to result in increased bioavailability, provided that the absorption of the drug is dissolution rate limited.
One of the classic examples of particle size effects on the bioavailability of poorly soluble compounds is that of griseofulvin, where a reduction of particle size from about 10 µm (specific surface area = 0.4 m2 g−1) to 2.7 µm (specific surface area = 1.5 m2 g−1) was shown to produce approximately double the amount of drug absorbed in humans. Many poorly soluble, slowly dissolving drugs are routinely presented in micronized form to increase their surface area.
Examples of drugs where a reduction in particle size has been shown to improve the rate and extent of oral absorption and hence bioavailability are shown in Table 20.2. Such improvements in bioavailability can result in an increased incidence of side-effects; thus for certain drugs it is important that the particle size is well controlled, and many pharmacopoeias state a requirement for particle size.
Table 20.2
Examples of drugs where a reduction in particle size has led to improvements in bioavailability
| Drug | Therapeutic class |
| Digoxin | Cardiac glycoside |
| Nitrofurantoin | Antifungal |
| Medroxyprogesterone | Hormone acetate |
| Danazol | Steroid |
| Tolbutamide | Antidiabetic |
| Aspirin | Analgesic |
| Sulfadiazine | Antibacterial |
| Naproxen | Non-steroidal anti-inflammatory |
| Ibuprofen | Non-steroidal anti-inflammatory |
| Phenacetin | Analgesic |
| Griseofulvin | Antifungal |
| Fenofibrate | Lipid regulating agent |
| Megestrol acetate | Apetite loss |
| Aprepitant | Anti-emetic |
| Rapamycin | Immunosuppressant |
| Lapinovir/ritonavir | HIV protease inhibitors |
For some drugs, particularly those that are hydrophobic in nature, micronization and other dry particle size reduction techniques can result in aggregation of the material. This will cause a consequent reduction in the effective surface area of the drug exposed to the gastrointestinal fluids and hence a reduction in its dissolution rate and bioavailability. Aspirin, phenacetin and phenobarbital are all prone to aggregation during particle size reduction. One approach that may overcome this problem is to micronize or mill the drug with a wetting agent or hydrophilic carrier. To overcome aggregation and to achieve particle sizes in the nano-size region, wet milling in the presence of stabilizers has been used. The relative bioavailability of danazol has been increased 400% by administering particles in the nanometre rather than the micrometre size range.
There are now several specialized drug delivery companies who can produce solid dosage forms with the drug stabilized in the nano-size range to afford greater bioavailaility. Examples of commercialized products are the immunosuppressant Rapamune®, the anti-emetic Emend® and the lipid regulating agent TriCor® containing fenofibrate. Megace® ES is an oral nano-suspension of megestrol acetate for the treatment of appetite loss, severe malnutrition, or unexplained significant weight loss in AIDS patients. It is a reformulation of the oral suspension using Nanocrystal® technology to improve the dissolution rate, absorption rate and bioavailability of the original formulation. The formulation is less viscous and allows a quarter of the volume to be dosed, thus aiding patient swallowing and compliance.
As well as milling with wetting agents, the effective surface area of hydrophobic drugs can be increased by the addition of a wetting agent to the formulation. The presence of polysorbate 80 in a fine suspension of phenacetin (particle size less than 75 µm) greatly improved the rate and extent of absorption of the phenacetin in human volunteers compared to the same-size suspension without a wetting agent. Polysorbate 80 helps by increasing the wetting and solvent penetration of the particles and by minimizing aggregation of suspended particles, thereby maintaining a large effective surface area. Wettability effects are highly drug specific; however wetting agents are routinely added to many formulations.
If an increase in the effective surface area of a drug does not increase its absorption rate, it is likely that the dissolution process is not rate limiting. For drugs such as penicillin G and erythromycin, which are unstable in gastric fluids, their chemical degradation will be minimized if they remain in the solid state. Thus, particle size reduction would not only serve to increase their dissolution rate but would simultaneously increase chemical degradation and therefore reduce the amount of intact drug available for absorption.
Solubility in the diffusion layer, Cs.
The dissolution rate of a drug under sink conditions, according to the Noyes–Whitney equation (Eqn 20.2), is directly proportional to its intrinsic solubility in the diffusion layer surrounding each dissolving drug particle, Cs. The aqueous solubility of a drug is dependent on the interactions between molecules within the crystal lattice, intermolecular interactions with the solution in which it is dissolving, and the entropy changes associated with fusion and dissolution. In the case of drugs that are weak electrolytes, their aqueous solubility is dependent on pH (as discussed in Chapter 2). Hence in the case of an orally administered solid dosage form containing a weak electrolyte drug, the dissolution rate of the drug will be influenced by its solubility and the pH in the diffusion layer surrounding each dissolving drug particle. The pH in the diffusion layer – the microclimate pH – for a weak electrolyte will be affected by the pKa and solubility of the dissolving drug and the pKa and solubility of the buffers in the bulk gastrointestinal fluids. Thus differences in dissolution rate will be expected in different regions of the gastrointestinal tract.
The solubility of weakly acidic drugs increases with pH and so as a drug moves down the gastrointestinal tract from the stomach to the intestine, its solubility will increase. Conversely, the solubility of weak bases decreases with increasing pH, i.e. as the drug moves down the gastrointestinal tract. It is important therefore for poorly soluble weak bases to dissolve rapidly in the stomach, as the rate of dissolution in the small intestine will be much slower. The antifungal drug ketoconazole, a weak base, is particularly sensitive to gastric pH. Dosing ketoconazole 2 hours after the administration of the H2 blocker cimetidine, which reduces gastric acid secretion, results in a significantly reduced rate and extent of absorption Similarly, in the case of the antiplatelet drug dipyrimidole, pretreatment with the H2 blocker famotidine reduces the peak plasma concentration by a factor of up to 10.
Salts.
The solubility of a weakly acidic drug in gastric fluid (pH 1–3.5) will be relatively low however it will be much greater in the higher pH of the intestine. The sodium salt of a weak acid will dissociate as shown:
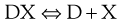 (20.3)
(20.3)
where D is the drug and X is the counter ion. The concentrations of the drug multiplied by the counter ion concentration at any pH will give the solubility product Ksp, i.e.:
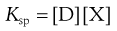 (20.4)
(20.4)
The pH solubility profile of a weak acid in the presence of counterions depends on the solubility product of the ionized drug and its counter-ions and is depicted for both a weak acid and a weak base.
Many examples can be found of the effects of salts improving the rate and extent of absorption. The dissolution rate of the oral hypoglycaemic drug, tolbutamide sodium in 0.1 M HC1 is 5000 times faster than that of the free acid. Oral administration of a non-disintegrating disc of the more rapidly dissolving sodium salt of tolbutamide produces a very rapid decrease in blood sugar level (a consequence of the rapid rate of drug absorption), followed by a rapid recovery. In contrast, a non-disintegrating disc of the tolbutamide free acid produces a much slower rate of decrease of blood sugar (a consequence of the slower rate of drug absorption) that is maintained over a longer period of time. The barbiturates are often administered in the form of sodium salts to achieve a rapid onset of sedation and provide more predictable effects.
The non-steroidal anti-inflammatory drug naproxen was originally marketed as the free acid for the treatment of rheumatoid and osteoarthritis. However, the sodium salt (naproxen sodium) is absorbed faster, due to faster dissolution of the dosage from, and hence is more effective, and thus has now largely replaced the free form. Conversely, strongly acidic salt forms of weakly basic drugs, for example chlorpromazine hydrochloride, dissolve more rapidly in gastric and intestinal fluids than do the free bases (e.g. chlorpromazine). The presence of strongly acidic anions (e.g. Cl− ions) in the diffusion layer around each drug particle ensures that the pH in that layer is lower than the bulk pH in either the gastric or the intestinal fluid. This lower pH will increase the solubility of the drug in the diffusion layer.
The oral administration of a salt form of a weakly basic drug in a solid oral dosage form generally ensures that dissolution occurs in the gastric fluid before the drug passes into the small intestine where pH conditions are unfavourable. Thus the drug should be delivered to the major absorption site, the small intestine, in solution. If absorption is fast enough, precipitation of the dissolved drug is unlikely to significantly affect bioavailability. It is important to be aware that hydrochloride salts may experience a common ion effect owing to the presence of chloride ions in the stomach (also discussed in Chapter 2). The in vitro dissolution of a sulfate salt of an HIV protease inhibitor analogue is significantly greater in hydrochloric acid than that of the hydrochloride salt. The bioavailability of the sulfate salt is more than three times greater than that of the hydrochloride salt. These observations are attributed to the common ion effect of the hydrochloride.
The sodium salts of acidic drugs and the hydrochloride salts of basic drugs are by far the most common. However, many other salt forms are increasingly being employed (there is further information in Chapter 23). Some salts have a lower solubility and dissolution rate than the free form, for example aluminium salts of weak acids and palmoate salts of weak bases. In these cases, insoluble films of either aluminium hydroxide or palmoic acid are found to coat the dissolving solids when the salts are exposed to a basic or an acidic environment, respectively. In general, poorly soluble salts delay absorption and may therefore be used to sustain the release of the drug. A poorly soluble salt form is generally employed for suspension dosage forms.
Although salt forms are often selected to improve bioavailability, other factors such as chemical stability, hygroscopicity, manufacturability and crystallinity will all be considered during salt selection and may preclude the choice of a particular salt. The sodium salt of aspirin, sodium acetylsalicylate, is much more prone to hydrolysis than is aspirin, acetylsalicylic acid, itself. One way to overcome chemical instabilities or other undesirable features of salts is to form the salt in situ or to add basic/acidic excipients to the formulation of a weakly acidic or weakly basic drug. The presence of the basic excipients in the formulation of acidic drugs ensures that a relatively basic diffusion layer is formed around each dissolving particle. The inclusion of the basic ingredients aluminium dihydroxyaminoacetate and magnesium carbonate in aspirin tablets was found to increase their dissolution rate and bioavailability.
Crystal form
Polymorphism
Many drugs can exist in more than one crystalline form. This property is referred to as polymorphism and each crystalline form is known as a polymorph (discussed further in Chapter 8). As discussed in Chapters 2 and 8, a metastable polymorph usually exhibits a greater dissolution rate than the corresponding stable polymorph. Consequently, the metastable polymorphic form of a poorly soluble drug may exhibit an increased bioavailability compared to the stable polymorphic form.
A classic example of the influence of polymorphism on drug bioavailability is provided by chloramphenicol palmitate. This drug exists in three crystalline forms designated A, B and C. At normal temperature and pressure, A is the stable polymorph, B is the metastable polymorph and C is the unstable polymorph. Polymorph C is too unstable to be included in a dosage form but polymorph B, the metastable form, is sufficiently stable. The plasma profiles of chloramphenicol from orally administered suspensions containing varying proportions of the polymorphic forms A and B were investigated. The extent of absorption of chloramphenicol increases as the proportion of the polymorphic form B of chloramphenicol palmitate is increased in each suspension. This was attributed to the more rapid in vivo rate of dissolution of the metastable polymorphic form, B, of chloramphenicol palmitate. Following dissolution, chloramphenicol palmitate is hydrolysed to give free chloramphenicol in solution, which is then absorbed. The stable polymorphic form A of chloramphenicol palmitate dissolves so slowly and consequently is hydrolysed so slowly to chloramphenicol in vivo that this polymorph is virtually ineffective. The importance of polymorphism to the gastrointestinal bioavailability of chloramphenicol palmitate is reflected by a limit being placed on the content of the inactive polymorphic form A, in chloramphenicol palmitate mixture.
Amorphous solids
In addition to different polymorphic crystalline forms, a drug may exist in an amorphous form (see Chapter 8). Because the amorphous form usually dissolves more rapidly than the corresponding crystalline form(s), the possibility exists that there will be significant differences in the bioavailabilities exhibited by the amorphous and crystalline forms of drugs that show dissolution rate-limited bioavailability.
A classic example of the influence of amorphous versus crystalline forms of a drug on its gastrointestinal bioavailability is provided by the antibiotic novobiocin. The more soluble and rapidly dissolving amorphous form of novobiocin was readily absorbed following oral administration of an aqueous suspension. However, the less soluble and slower dissolving crystalline form was not absorbed to any significant extent. The crystalline form was thus therapeutically ineffective. A further important observation was made in the case of aqueous suspensions of novobiocin. The amorphous form slowly converts to the more thermodynamically stable crystalline form, with an accompanying loss of therapeutic effectiveness. Thus, unless adequate precautions are taken to ensure the stability of the less stable, more therapeutically effective amorphous form of a drug in a dosage form, then unacceptable variations in therapeutic effectiveness may occur.
Several delivery technologies for poorly soluble drugs rely on stabilizing the drug in its amorphous form to increase its dissolution and bioavailability. An example of this is Kaletra® which is a combination tablet of the protease inhibitors lopinavir and ritinovir used for HIV infection, in combination with other antiretroviral drugs. These drugs are stabilized in their amorphous form by a polymer, copovidone, following melt extrusion of the drug with the polymer. The tablets provide a significant improvement in bioavailability and variability such that two medium sized tablets are equivalent to three large capsules of the old formulation.
Solvates
Another variation in the crystalline form of a drug can occur if the drug is able to associate with solvent molecules to produce crystalline forms known as solvates (discussed further in Chapter 8). When water is the solvent, the solvate formed is called a hydrate. Generally, the greater the solvation of the crystal, the lower is the solubility and dissolution rate in a solvent identical to the solvation molecules. As the solvated and non-solvated forms usually exhibit differences in dissolution rates, they may also exhibit differences in bioavailability, particularly in the case of poorly soluble drugs that exhibit dissolution rate-limited bioavailability.
An example is that of the antibiotic ampicillin. The faster dissolving anhydrous form of ampicillin is absorbed to a greater extent from both hard gelatin capsules and an aqueous suspension than is the slower dissolving trihydrate form. The anhydrous form of the hydrochloride salt of a protease inhibitor, an analogue of indinavir, has a much faster dissolution rate than the hydrated form in water. This is reflected in a significantly greater rate and extent of absorption and a more than doubling of the bioavailability of the anhydrous form.
Factors affecting the concentration of drug in solution in the gastrointestinal fluids
The rate and extent of absorption of a drug depend on the effective concentration of that drug, i.e. the concentration of drug in solution in the gastrointestinal fluids, which is in an absorbable form. Complexation, micellar solubilization, adsorption and chemical stability are the principal physicochemical properties that can influence the effective drug concentration in the gastrointestinal fluids.
Complexation.
Complexation of a drug may occur within the dosage form and/or in the gastrointestinal fluids, and can be beneficial or detrimental to absorption.
Mucin which is present in gastrointestinal fluids forms complexes with some drugs. The antibiotic streptomycin binds to mucin, thereby reducing the available concentration of the drug for absorption. It is thought that this may contribute to its poor bioavailability. Another example of complexation is that between drugs and dietary components, as in the case of the tetracyclines, which is discussed in Chapter 19.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


