• Associated extrahepatic congenital anomalies present in up to 20% of cases
• Surgical intervention required
Kasai procedure to reestablish bile flow; best outcome if performed before 45-60 days of age
Most frequent indication for pediatric liver transplantation
Macroscopic
• Level of extrahepatic duct obliteration is most common within portal hepatis
• Associated with hypoplastic or atretic gallbladder
Microscopic
• Ductular reaction, duct/ductular bile plugs, and portal and periportal fibrosis
• Associated with lobular cholestasis, focal giant cell transformation, and extramedullary hematopoiesis
• Careful clinical and radiographic correlation required to exclude other entities in differential diagnosis
Top Differential Diagnoses
• Idiopathic neonatal hepatitis
• α-1-antitrypsin deficiency
• Total parenteral nutrition-associated cholestasis
• CMV infection
• Choledochal cyst
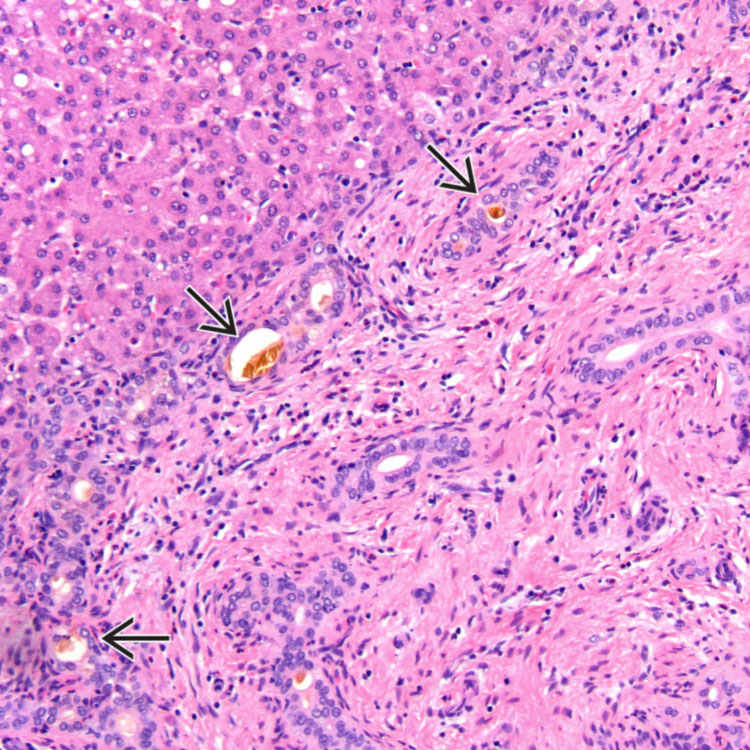
Portal Expansion and Ductular Reaction Wedge liver biopsy specimen from an 8-week-old infant shows an expanded portal tract with ductular reaction. The ductules along the periphery contain bile plugs .
Portal Expansion Trichrome stain highlights the expanded portal tracts with associated ductular reaction in a liver biopsy specimen from a 7-week-old infant.
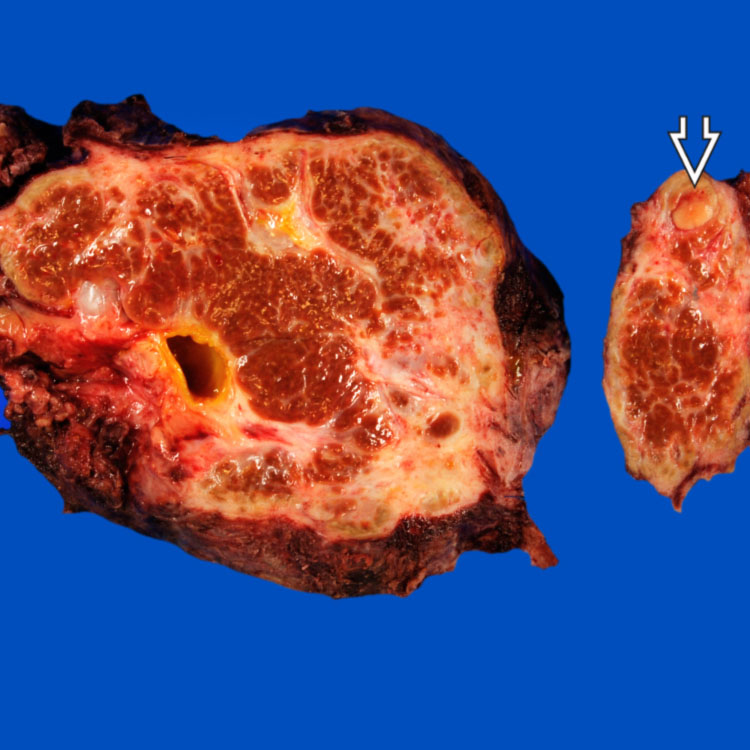
Gross Appearance Explanted liver from a 12 year old with biliary atresia (BA) is cirrhotic. In the left lobe of the liver, a 0.9-cm yellow nodule was discovered. Histologically, this was a well-differentiated hepatocellular carcinoma.
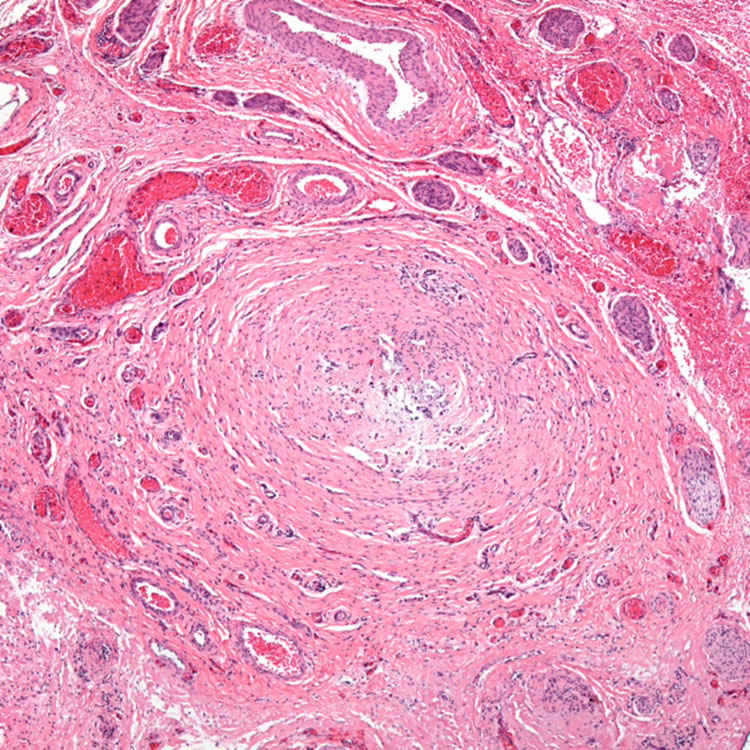
Biliary Remnant Section of biliary remnant removed during Kasai procedure demonstrates a markedly narrowed bile duct-like structure with a nearly absent lumen surrounded by fibrosis. Distal to this section, a patent bile duct lumen was identified.
TERMINOLOGY
Abbreviations
• Biliary atresia (BA)
Synonyms
• Extrahepatic biliary atresia
Involves both extrahepatic and intrahepatic biliary tree
– Thus, best classified simply as BA
Definitions
• Idiopathic fibroinflammatory process that destroys bile ducts
May culminate in ductopenia and biliary cirrhosis
ETIOLOGY/PATHOGENESIS
Idiopathic
• Probable multiple disease mechanisms
Possible roles for viral infection, genetic factors, congenital malformations
Associated genetic/chromosomal abnormalities
– Trisomy 18 and 21
– Cateye and Kabuki syndromes
CLINICAL ISSUES
Epidemiology
• Incidence
1:5,000 to 1:19,000 newborns
Most common in East Asian countries
Presentation
• Most common cause of pathologic infant jaundice
• Clinical triad
Persistent neonatal jaundice, beyond 2 weeks of life
Dark urine and acholic pale stools
Hepatomegaly
• Associated extrahepatic congenital anomalies present in up to 20% of cases
Most common is biliary atresia splenic malformation syndrome
• 2 general clinical patterns
Prenatal, embryonal/fetal, congenital, or early form
– 15-35% of cases
– Low birth weight, jaundice at birth
Perinatal, postnatal, infantile, acquired, or late form
– 65-85% of cases
– Healthy anicteric, average-weight neonates
– Jaundice usually presents after 2 weeks of age
Laboratory Tests
• Similar to other forms of neonatal cholestasis
Conjugated hyperbilirubinemia
Variably elevated alkaline phosphatase
Variably elevated transaminases
γ-glutamyl transpeptidase typically > 200 U/L
Treatment
• Surgery
Hepatoportoenterostomy (Kasai procedure)
– Palliative procedure to reestablish some bile flow
– Best if performed before 45-60 days of age
Liver transplantation
– Biliary atresia most frequent indication for pediatric liver transplantation
– For infants without bile drainage procedure, transplant within 6 months to 2 years of age
Prognosis
• Fatal by age 2 if untreated
Prenatal form has worse outcome than postnatal form
Only gold members can continue reading. Log In or Register to continue



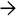 .
.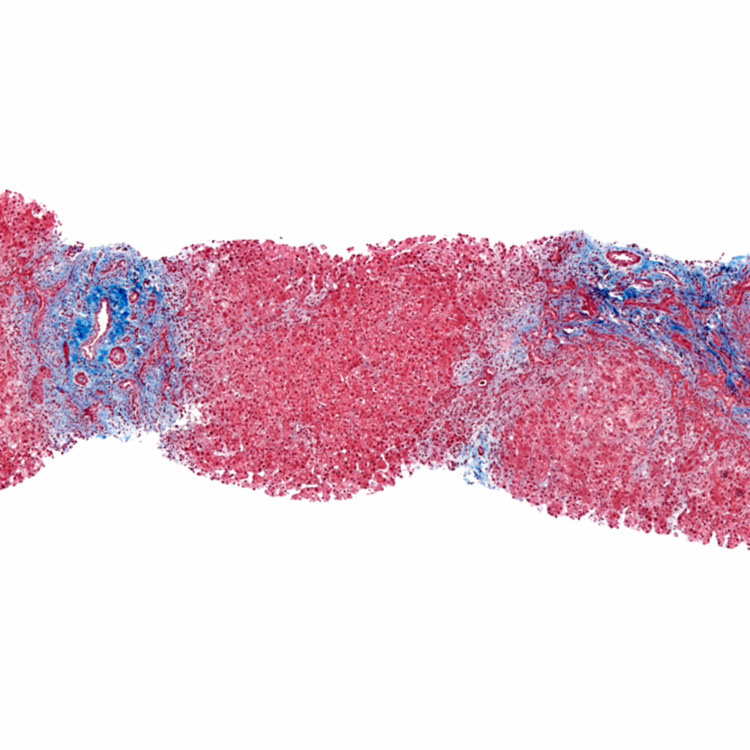
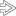 was discovered. Histologically, this was a well-differentiated hepatocellular carcinoma.
was discovered. Histologically, this was a well-differentiated hepatocellular carcinoma.