Autoimmune Lymphoproliferative Syndrome
Sa A. Wang, MD
Key Facts
Etiology/Pathogenesis
Disease of disrupted lymphocyte homeostasis as result of defective Fas-mediated apoptosis
Many mutations have been identified in ALPS
Type I: Accounts for approximately 65% of all ALPS cases; 3 subtypes
Ia: Germline mutations in FAS gene
Ib: Germline mutations in FAS ligand gene
Is: Somatic mutations in FAS gene
Type II: Germline mutations in gene encoding caspase 10
Type III: Accounts for approximately 20-30% of all ALPS cases
No identifiable genetic mutations in FAS pathway
Type IV: Very rare; gain-of-function mutation in NRAS
Clinical Issues
Chronic nonmalignant lymphoproliferation
Lymph nodes, spleen, liver
Autoimmune disease
Increased risk for lymphoma
Microscopic Pathology
Marked paracortical expansion with increased DNT cells
Ancillary Tests
Flow cytometry
Increased DNT cells: TCR-α/β(+), CD3(+), CD4(-), CD8(-)
Apoptosis assay: Defective FAS-induced apoptosis in ALPS types Ia and Ib
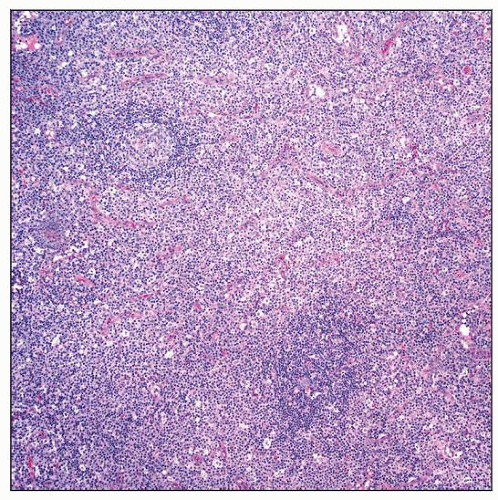 Autoimmune lymphoproliferative syndrome (ALPS) involving lymph node. The paracortex of the lymph node is markedly expanded. Small lymphoid follicles are also present. |
 ALPS involving lymph node. The paracortex is populated by small lymphocytes and many large immunoblasts with prominent nucleoli. |
TERMINOLOGY
Abbreviations
Autoimmune lymphoproliferative syndrome (ALPS)
Definitions
Disease of disrupted lymphocyte homeostasis as result of defective Fas-mediated apoptosis
ETIOLOGY/PATHOGENESIS
Genetic Mutations in FAS Pathway
FAS pathway mutations cause ALPS
FAS mutations are usually heterozygous
Multiple types have been described
Type I: Accounts for approximately 65% of all ALPS cases
3 type I subtypes
Ia: Germline mutations in FAS (TNFRSF6, CD95, APO1) gene
Ib: Germline mutations in FAS ligand gene
Is: Somatic mutations in FAS gene
Type II: Germline mutations in gene encoding caspase 10
Type III: No identifiable genetic mutations in FAS pathway
Accounts for approximately 20-30% of all ALPS cases
Type IV: Very rare
Gain-of-function mutation in NRAS
Patients have ALPS phenotype but normal Fas-mediated apoptosis
ALPS is multistep process requiring more than a single genetic hit for clinical expression
In most cases, mutations are inherited in autosomal dominant fashion
Therefore, penetrance is 100% at cellular level
Penetrance for clinical phenotype of ALPS is variable
Significant proportion of family members can have mutation without phenotype of ALPS
Additional factors must contribute to expression of disease
CASPASE 8 Mutations
Once considered part of ALPS
Present with lymphadenopathy and defective Fas-mediated apoptosis
Profound apoptotic defects in B, T, and NK cells
Patients often have mucocutaneous herpes virus infections
Therefore, CASP8 mutations are now considered to represent a distinct disease
CLINICAL ISSUES
Presentation
Chronic nonmalignant lymphoproliferation, often appearing in 1st year of life
Chronic &/or recurrent lymphadenopathy in ˜ 80% of patients
Splenomegaly with/without hypersplenism in ˜ 85% of patients
Hepatomegaly in ˜ 45% of patients
Lymphocytic interstitial pneumonia
Autoimmune diseases in ˜ 70% of patients
Cytopenias are most frequent
Autoimmune hemolytic anemia
Immune thrombocytopenia
Autoimmune neutropenia
More than 1 lineage is often affected
Evans syndrome
Originally described in 1951
Autoimmune destruction of erythrocytes and platelets
Subset of these patients has ALPS
Other less common autoimmune phenomena in ALPS include
Skin rash: Often of urticarial nature
Autoimmune hepatitis
Autoimmune glomerulonephritis
Autoimmune thyroiditis
Uveitis and Guillain-Barré syndrome
Vasculitis and panniculitis
Autoimmune colitis
Autoimmune cerebellar syndrome
Patients followed into adulthood have increased risk of pulmonary fibrosis
ALPS patients have increased risk of malignancies of various types
Increased risk of Hodgkin lymphoma and non-Hodgkin lymphoma
51x increased risk of Hodgkin lymphoma
14x increased risk of non-Hodgkin lymphoma
Usually not related to Epstein-Barr virus infection
Increased risk of carcinomas
Thyroid, breast, liver, tongue, skin
Increased risk of leukemias
Some ALPS patients present with multiple neoplasms (thyroid/breast adenomas, gliomas)
Presentation related to type of genetic mutation
Homozygous or compound heterozygous FAS mutations lead to
Severe lymphoproliferation before, at, or shortly after birth
Patients typically succumb to lymphoproliferation &/or autoimmunity at early age
Mutations in any domain of Fas lead to same clinical phenotype of ALPS
Lymphoma is most often associated with mutations affecting intracellular domains of Fas
Laboratory Tests
Peripheral blood lymphocytosis
Serum
Elevated concentrations of IgG, IgA, and IgE; normal or decreased concentration of IgM
Increased levels of interleukin (IL)-10
Increased levels of vitamin B12
Autoimmune antibodies
Autoantibodies to red cells, platelets, and neutrophils are often found
Anti-smooth muscle and anti-phospholipid antibodies can be positive
Anti-nuclear antibodies and rheumatoid factor can be positive
Flow cytometric immunophenotyping of peripheral blood shows increased double negative T cells
Double negative T cells (DNT) = TCR-α/β(+), CD3(+), CD4(-), CD8(-)
Normal range: DNT cells have been expressed as percentage of total lymphocytes; total T cells and TCR-α/β(+) T cells in various studies
Normal range may differ according to patient age and flow cytometry gating strategy
DNT cells are increased if > 1% of total T cells (peripheral blood)
Markedly increased (3-60%) DNTs in peripheral blood is very specific for ALPS
Present in all subtypes of ALPS
Found in peripheral blood, lymph nodes, spleen, and other tissues
Role of DNT cells in ALPS, and whether these cells are pathogenic or merely a marker of disease, remains to be determined
Other flow cytometry findings
Increased TCR-γ/δ(+) DNT cells
Increased CD8(+), CD57(+) T cells
Increased CD5(+) B cells
Increased HLA-DR(+) T cells
Decreased CD27(+) B cells
Decreased CD4(+), CD25(+) regulatory T cells
DNT can be increased in other autoimmune diseases
Usually low-level increase of DNT in these diseases
Systemic lupus erythematosus
Immune thrombocytopenic purpura
FAS mutations in 100% of DNT population in somatic ALPS patients suggests that these cells contribute to disease pathogenesis
In vitro Fas-mediated apoptosis assays are helpful for diagnosis of ALPS
Isolate peripheral blood mononuclear cells from ALPS patient
Activate T cells with mitogen and expand with IL-2 in culture for 28 days
Expose T cells to anti-Fas IgM antibody
Normal T cells: Rapid cell death and apoptosis
ALPS T cells: No or impaired cell death
Type of ALPS mutation yields different results for in vitro Fas-mediated apoptosis
Type I: Often exhibit defective FAS-induced apoptosis
Types II and III: No defective FAS-induced apoptosis
Molecular genetic assays
FAS
FAS germline mutations identified throughout entire coding region and exons/introns of FAS
Sequencing of entire coding region and intron/exon boundaries of FAS gene detects ˜ 90% of mutations
FAS somatic mutation detection often performed on sorted DNT cells
FASLG
Sequence analysis of entire coding region of FASLG gene is available clinically
CASP10
Sequence analysis of entire coding region of CASP10 gene is available clinically
Natural History
Nonmalignant lymphoproliferative manifestations in ALPS often regress or improve over time
Autoimmunity shows no permanent remission with advancing age
Risk for development of lymphoma appears to be lifelong
Treatment
Some patients with ALPS require no treatment
Hemolytic anemia and thrombocytopenia
Prednisone
Immunosuppressant
Mycophenolate mofetil (Cellcept)
Sirolimus (rapamycin)
Only a few patients respond to intravenous immunoglobulin
Rituximab: Anti-CD20 monoclonal chimeric antibody
Percentage of ALPS patients are predisposed to develop common variable immunodeficiency disease (CVID) upon rituximab treatment
Reserved for patients who fail all other therapies
Splenectomy to control autoimmune cytopenias is discouraged
ALPS patients have increased risk of developing post-splenectomy sepsis despite vaccination and antibiotic prophylaxis
No long-term effect to control cytopenia(s)
Bone marrow (hematopoietic stem cell) transplantation carries risks
Reduced-intensity transplant can reduce transplant-associated risks
Prognosis
Refer to natural history
Recently Proposed Diagnostic Criteria for ALPS
Major
1: Chronic nonmalignant lymphoproliferation
> 6 months
Splenomegaly &/or lymphadenopathy of at least 2 nodal groups
2: Marked elevation of peripheral blood DNTs of at least 5%
3: Defective in vitro Fas-mediated apoptosis
4: Identifiable genetic mutation, germline or somatic
FAS, FASL, CASP10, NRAS
Minor
1: Autoimmune cytopenias
Thrombocytopenia, neutropenia, &/or hemolytic anemia
Proven to be immune-mediated by autoantibody detection or response to immunosuppressive agent
2: Moderate elevation in DNTs
3: Elevated serum IgG
4: Elevated serum IL-10
5: Elevated serum vitamin B12
6: Elevated plasma Fas ligand level
Diagnosis established if
3 major criteria present or
2 major + 2 minor criteria present
IMAGE FINDINGS
Radiographic Findings
Imaging studies detect lymphadenopathy or hepatosplenomegaly
Lymphoproliferations in ALPS are FDG PET avid
Cannot distinguish benign from malignant; therefore, biopsy needed
MICROSCOPIC PATHOLOGY
Lymph Nodes
Marked expansion of paracortical (T-cell) zones
Lymphocytes show various stages of immunoblastic transformation
Small, intermediate, and large lymphocytes; often with clear cytoplasm
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


