363 | Alcoholic Liver Disease |
Chronic and excessive alcohol ingestion is one of the major causes of liver disease. The pathology of alcoholic liver disease consists of three major lesions, with the progressive injury rarely existing in a pure form: (1) fatty liver, (2) alcoholic hepatitis, and (3) cirrhosis. Fatty liver is present in >90% of daily as well as binge drinkers. A much smaller percentage of heavy drinkers will progress to alcoholic hepatitis, thought to be a precursor to cirrhosis. The prognosis of severe alcoholic liver disease is dismal; the mortality of patients with alcoholic hepatitis concurrent with cirrhosis is nearly 60% at 4 years. Although alcohol is considered a direct hepatotoxin, only between 10 and 20% of alcoholics will develop alcoholic hepatitis. The explanation for this apparent paradox is unclear but involves the complex interaction of facilitating factors, such as drinking patterns, diet, obesity, and gender. There are no diagnostic tools that can predict individual susceptibility to alcoholic liver disease.
GLOBAL CONSIDERATIONS
![]() Alcohol is the world’s third largest risk factor for disease burden. The harmful use of alcohol results in 2.5 million deaths each year. Most of the mortality attributed to alcohol is secondary to cirrhosis. Mortality from cirrhosis is declining in most Western countries, concurrent with a reduction in alcohol consumption, with the exceptions of the United Kingdom, Russia, Romania, and Hungary. These increases in cirrhosis and its complications are closely correlated with increased volume of alcohol consumed per capita population and are regardless of gender.
Alcohol is the world’s third largest risk factor for disease burden. The harmful use of alcohol results in 2.5 million deaths each year. Most of the mortality attributed to alcohol is secondary to cirrhosis. Mortality from cirrhosis is declining in most Western countries, concurrent with a reduction in alcohol consumption, with the exceptions of the United Kingdom, Russia, Romania, and Hungary. These increases in cirrhosis and its complications are closely correlated with increased volume of alcohol consumed per capita population and are regardless of gender.
ETIOLOGY AND PATHOGENESIS
Quantity and duration of alcohol intake are the most important risk factors involved in the development of alcoholic liver disease (Table 363-1). The roles of beverage type(s), i.e. wine, beer, or spirits, and pattern of drinking (daily versus binge drinking) are less clear. Progress beyond the fatty liver stage seems to require additional risk factors that remain incompletely defined. Although there are genetic predispositions for alcoholism (Chap. 467), gender is a strong determinant for alcoholic liver disease. Women are more susceptible to alcoholic liver injury when compared to men. They develop advanced liver disease with substantially less alcohol intake. In general, the time it takes to develop liver disease is directly related to the amount of alcohol consumed. It is useful in estimating alcohol consumption to understand that one beer, four ounces of wine, or one ounce of 80% spirits all contain ~12 g of alcohol. The threshold for developing alcoholic liver disease is higher in men, while women are at increased risk for developing similar degrees of liver injury by consuming significantly less. Gender-dependent differences result from poorly understood effects of estrogen, proportion of body fat, and the gastric metabolism of alcohol. Obesity, a high-fat diet, and the protective effect of coffee have been postulated to play a part in the development of the pathogenic process.
RISK FACTORS FOR ALCOHOLIC LIVER DISEASE |

Chronic infection with hepatitis C virus (HCV) (Chap. 362) is an important comorbidity in the progression of alcoholic liver disease to cirrhosis in chronic and excessive drinkers. Even moderate alcohol intake of 20–50 g/d increases the risk of cirrhosis and hepatocellular cancer in HCV-infected individuals. Patients with both alcoholic liver injury and HCV infection develop decompensated liver disease at a younger age and have poorer overall survival. Increased liver iron stores and, rarely, porphyria cutanea tarda can occur as a consequence of the overlapping injurious processes secondary to alcohol abuse and HCV infection. In addition, alcohol intake of >50 g/d by HCV-infected patients decreases the efficacy of interferon-based antiviral therapy.
The pathogenesis of alcoholic liver injury is unclear. The present conceptual foundation is that alcohol acts as a direct hepatotoxin and that malnutrition does not have a major role. Ingestion of alcohol initiates an inflammatory cascade by its metabolism to acetaldehyde, resulting in a variety of metabolic responses. Steatosis from lipogenesis, fatty acid synthesis, and depression of fatty acid oxidation appears secondary to effects on sterol regulatory transcription factor and peroxisome proliferator-activated receptor α (PPAR-α). Intestinal-derived endotoxin initiates a pathogenic process through toll-like receptor 4 and tumor necrosis factor α (TNF-α) that facilitates hepatocyte apoptosis and necrosis. The cell injury and endotoxin release initiated by ethanol and its metabolites also activate innate and adaptive immunity pathways releasing proinflammatory cytokines (e.g., TNF-α), chemokines, and proliferation of T and B cells. The production of toxic protein-aldehyde adducts, generation of reducing equivalents, and oxidative stress also contribute to the liver injury. Hepatocyte injury and impaired regeneration following chronic alcohol ingestion are ultimately associated with stellate cell activation and collagen production, which are key events in fibrogenesis. The resulting fibrosis from continuing alcohol use determines the architectural derangement of the liver and associated pathophysiology.
PATHOLOGY
The liver has a limited repertoire in response to injury. Fatty liver is the initial and most common histologic response to hepatotoxic stimuli, including excessive alcohol ingestion. The accumulation of fat within the perivenular hepatocytes coincides with the location of alcohol dehydrogenase, the major enzyme responsible for alcohol metabolism. Continuing alcohol ingestion results in fat accumulation throughout the entire hepatic lobule. Despite extensive fatty change and distortion of the hepatocytes with macrovesicular fat, the cessation of drinking results in normalization of hepatic architecture and fat content. Alcoholic fatty liver has traditionally been regarded as entirely benign, but similar to the spectrum of nonalcoholic fatty liver disease (Chap. 367e), the appearance of steatohepatitis and certain pathologic features such as giant mitochondria, perivenular fibrosis, and macrovesicular fat may be associated with progressive liver injury.
The transition between fatty liver and the development of alcoholic hepatitis is blurred. The hallmark of alcoholic hepatitis is hepatocyte injury characterized by ballooning degeneration, spotty necrosis, polymorphonuclear infiltrate, and fibrosis in the perivenular and perisinusoidal space of Disse. Mallory-Denk bodies are often present in florid cases but are neither specific nor necessary to establish the diagnosis. Alcoholic hepatitis is thought to be a precursor to the development of cirrhosis. However, like fatty liver, it is potentially reversible with cessation of drinking. Cirrhosis is present in up to 50% of patients with biopsy-proven alcoholic hepatitis, and its regression is uncertain, even with abstention.
CLINICAL FEATURES
The clinical manifestations of alcoholic fatty liver are subtle and characteristically detected as a consequence of the patient’s visit for a seemingly unrelated matter. Previously unsuspected hepatomegaly is often the only clinical finding. Occasionally, patients with fatty liver will present with right upper quadrant discomfort, nausea, and, rarely, jaundice. Differentiation of alcoholic fatty liver from nonalcoholic fatty liver is difficult unless an accurate drinking history is ascertained. In every instance where liver disease is present, a thoughtful and sensitive drinking history should be obtained. Standard, validated questions accurately detect alcohol-related problems (Chap. 467). Alcoholic hepatitis is associated with a wide gamut of clinical features. Fever, spider nevi, jaundice, and abdominal pain simulating an acute abdomen represent the extreme end of the spectrum, while many patients will be entirely asymptomatic. Portal hypertension, ascites, or variceal bleeding can occur in the absence of cirrhosis. Recognition of the clinical features of alcoholic hepatitis is central to the initiation of an effective and appropriate diagnostic and therapeutic strategy. It is important to recognize that patients with alcoholic cirrhosis often exhibit clinical features identical to other causes of cirrhosis.
LABORATORY FEATURES
Patients with alcoholic liver disease are often identified through routine screening tests. The typical laboratory abnormalities seen in fatty liver are nonspecific and include modest elevations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and γ-glutamyl transpeptidase (GGTP), often accompanied by hypertriglyceridemia and hyperbilirubinemia. In alcoholic hepatitis and in contrast to other causes of fatty liver, AST and ALT are usually elevated two- to sevenfold. They are rarely >400 IU, and the AST/ALT ratio is >1 (Table 363-2). Hyperbilirubinemia is accompanied by modest increases in the alkaline phosphatase level. Derangement in hepatocyte synthetic function indicates more serious disease. Hypoalbuminemia and coagulopathy are common in advanced liver injury. Ultrasonography is useful in detecting fatty infiltration of the liver and determining liver size. The demonstration by ultrasound of portal vein flow reversal, ascites, and intraabdominal venous collaterals indicates serious liver injury with less potential for complete reversal.
LABORATORY DIAGNOSIS OF ALCOHOLIC FATTY LIVER AND ALCOHOLIC HEPATITIS |

PROGNOSIS
Critically ill patients with alcoholic hepatitis have short-term (30-day) mortality rates >50%. Severe alcoholic hepatitis is heralded by coagulopathy (prothrombin time increased >5 s), anemia, serum albumin concentrations <25 g/L (2.5 mg/dL), serum bilirubin levels >137 μmol/L (8 mg/dL), renal failure, and ascites. A discriminant function calculated as 4.6 × (the prolongation of the prothrombin time above control [seconds]) + serum bilirubin (mg/dL) can identify patients with a poor prognosis (discriminant function >32). A Model for End-Stage Liver Disease (MELD) score (Chap. 368) ≥21 also is associated with significant mortality in alcoholic hepatitis. The presence of ascites, variceal hemorrhage, deep encephalopathy, or hepatorenal syndrome predicts a dismal prognosis. The pathologic stage of the injury can be helpful in predicting prognosis. Liver biopsy should be performed whenever possible to establish the diagnosis and to guide the therapeutic decisions.
TREATMENT | ALCOHOLIC LIVER DISEASE |
Complete abstinence from alcohol is the cornerstone in the treatment of alcoholic liver disease. Improved survival and the potential for reversal of histologic injury regardless of the initial clinical presentation are associated with total avoidance of alcohol ingestion. Referral of patients to experienced alcohol counselors and/or alcohol treatment programs should be routine in the management of patients with alcoholic liver disease. Attention should be directed to the nutritional and psychosocial states during the evaluation and treatment periods. Because of data suggesting that the pathogenic mechanisms in alcoholic hepatitis involve cytokine release and the perpetuation of injury by immunologic processes, glucocorticoids have been extensively evaluated in the treatment of alcoholic hepatitis. Patients with severe alcoholic hepatitis, defined as a discriminant function >32 or MELD >20, should be given prednisone, 40 mg/d, or prednisolone, 32 mg/d, for 4 weeks, followed by a steroid taper (Fig. 363-1). Exclusion criteria include active gastrointestinal bleeding, renal failure, or pancreatitis. Women with encephalopathy from severe alcoholic hepatitis may be particularly good candidates for glucocorticoids. A Lille score >0.45, at http://www.lillemodel.com, uses pretreatment variables plus the change in total bilirubin at day 7 of glucocorticoids to identify patients unresponsive to therapy.
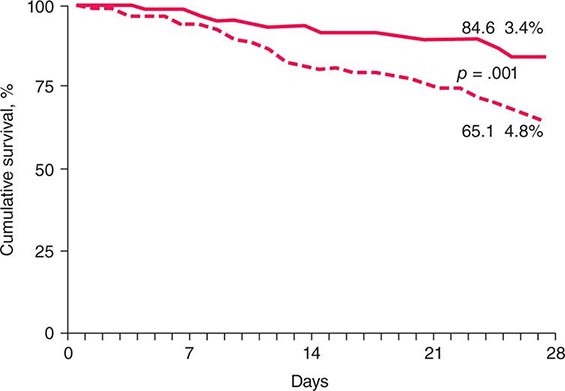
FIGURE 363-1 Effect of glucocorticoid therapy of severe alcoholic hepatitis on short-term survival: the result of a meta-analysis of individual data from three studies. Prednisolone, solid line; placebo, dotted line. (Adapted from P Mathurin et al: J Hepatol 36:480, 2002, with permission from Elsevier Science.)
The role of TNF-α expression and receptor activity in alcoholic liver injury has led to an examination of TNF inhibition as an alternative to glucocorticoids for severe alcoholic hepatitis. The nonspecific TNF inhibitor, pentoxifylline, demonstrated improved survival in the therapy of severe alcoholic hepatitis, primarily due to a decrease in hepatorenal syndrome (Fig. 363-2). Monoclonal antibodies that neutralize serum TNF-α should not be used in alcoholic hepatitis because of studies reporting increased deaths secondary to infection and renal failure.
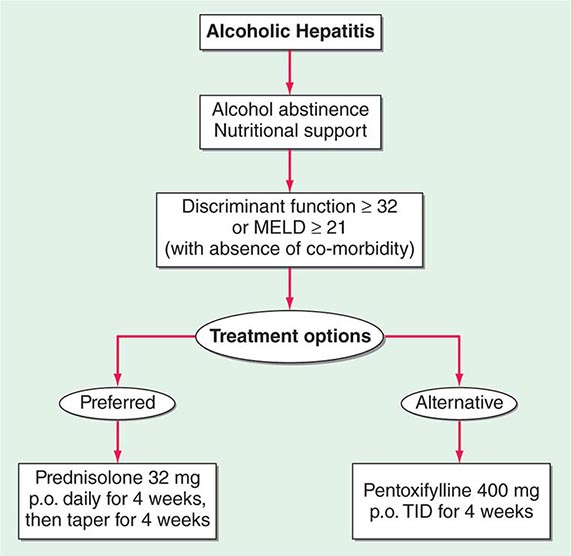
FIGURE 363-2 Treatment algorithm for alcoholic hepatitis. As identified by a calculated discriminant function >32 (see text), patients with severe alcoholic hepatitis, without the presence of gastrointestinal bleeding or infection, would be candidates for either glucocorticoids or pentoxifylline administration.
Liver transplantation is an accepted indication for treatment in selected and motivated patients with end-stage cirrhosis. Outcomes are equal or superior to other indications for transplantation. In general, transplant candidacy should be reevaluated after a defined period of sobriety. Patients presenting with alcoholic hepatitis have been largely excluded from transplant candidacy because of the perceived risk of increased surgical mortality and high rates of recidivism following transplantation. Recently, a European multidisciplinary group has reported excellent long-term transplant outcomes in highly selected patients with florid alcoholic hepatitis. General application of transplantation in such patients must await confirmatory outcomes by others.
364 | Nonalcoholic Fatty Liver Diseases and Nonalcoholic Steatohepatitis |
INCIDENCE, PREVALENCE, AND NATURAL HISTORY
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in many parts of the world, including the United States. Population-based abdominal imaging studies have demonstrated fatty liver in at least 25% of American adults. Because the vast majority of these subjects deny hazardous levels of alcohol consumption (defined as greater than one drink per day in women or two drinks per day in men), they are considered to have NAFLD. NAFLD is strongly associated with overweight/obesity and insulin resistance. However, it can also occur in lean individuals and is particularly common in those with a paucity of adipose depots (i.e., lipodystrophy). Ethnic/racial factors also appear to influence liver fat accumulation; the documented prevalence of NAFLD is lowest in African Americans (~25%), highest in Americans of Hispanic ancestry (~50%), and intermediate in American whites (~33%).
NAFLD encompasses a spectrum of liver pathology with different clinical prognoses. The simple accumulation of triglyceride within hepatocytes (hepatic steatosis) is on the most clinically benign extreme of the spectrum. On the opposite, most clinically ominous extreme, are cirrhosis (Chap. 365) and primary liver cancer (Chap. 111). The risk of developing cirrhosis is extremely low in individuals with chronic hepatic steatosis, but increases as steatosis becomes complicated by histologically conspicuous hepatocyte death and inflammation (i.e., nonalcoholic steatohepatitis [NASH]). NASH itself is also a heterogeneous condition; sometimes it improves to steatosis or normal histology, sometimes it remains relatively stable for years, but sometimes it results in progressive accumulation of fibrous scar that eventuates in cirrhosis. Once NAFLD-related cirrhosis develops, the annual incidence of primary liver cancer is 1%.
Abdominal imaging is not able to determine which individuals with NAFLD have associated liver cell death and inflammation (i.e., NASH), and specific blood tests to diagnose NASH are not yet available. However, population-based studies that have used elevated serum ALT as a marker of liver injury indicate that about 6–8% of American adults have serum ALT elevations that cannot be explained by excessive alcohol consumption, other known causes of fatty liver disease (Table 364-1), viral hepatitis, or drug-induced or congenital liver diseases. Because the prevalence of such “cryptogenic” ALT elevations increases with body mass index, it is presumed that they are due to NASH. Hence, at any given point in time, NASH is present in about 25% of individuals who have NAFLD (i.e., about 6% of the general U.S. adult population has NASH). Smaller cross-sectional studies in which liver biopsies have been performed on NASH patients at tertiary referral centers consistently demonstrate advanced fibrosis or cirrhosis in about 25% of those cohorts. By extrapolation, therefore, cirrhosis develops in about 6% of individuals with NAFLD (i.e., in about 1.5–2% of the general U.S. population). The risk for advanced liver fibrosis is highest in individuals with NASH who are older than 45–50 years of age and overweight/obese or afflicted with type 2 diabetes.
ALTERNATIVE CAUSES OF HEPATIC STEATOSIS |
To put these data in perspective, it is helpful to recall that the prevalence of hepatitis C–related cirrhosis in the United States is about 0.5%. Thus, NAFLD-related cirrhosis is about three to four times more common than cirrhosis caused by chronic hepatitis C infection. Consistent with these data, experts have predicted that NAFLD will surpass hepatitis C as the leading indication for liver transplantation in the United States within the next decade. Similar to cirrhosis caused by other liver diseases, cirrhosis caused by NAFLD increases the risk for primary liver cancer. Both hepatocellular carcinoma and intrahepatic cholangiocarcinoma (ICC) have also been reported to occur in NAFLD patients without cirrhosis, suggesting that NAFLD per se may be a premalignant condition. NAFLD, NASH, and NAFLD-related cirrhosis are not limited to adults. All have been well documented in children. As in adults, obesity and insulin resistance are the main risk factors for pediatric NAFLD. Thus, the rising incidence and prevalence of childhood obesity suggests that NAFLD is likely to become an even greater contributor to society’s burden of liver disease in the future.
PATHOGENESIS
The mechanisms underlying the pathogenesis and progression of NAFLD are not entirely clear. The best-understood mechanisms pertain to hepatic steatosis. This is proven to result when hepatocyte mechanisms for triglyceride synthesis (e.g., lipid uptake and de novo lipogenesis) overwhelm mechanisms for triglyceride disposal (e.g., degradative metabolism and lipoprotein export), leading to accumulation of fat (i.e., triglyceride) within hepatocytes. Obesity stimulates hepatocyte triglyceride accumulation by altering the intestinal microbiota to enhance both energy harvest from dietary sources and intestinal permeability. Reduced intestinal barrier function increases hepatic exposure to gut-derived products, which stimulate liver cells to generate inflammatory mediators that inhibit insulin actions. Obese adipose depots also produce excessive soluble factors (adipokines) that inhibit tissue insulin sensitivity. Insulin resistance promotes hyperglycemia. This drives the pancreas to produce more insulin to maintain glucose homeostasis. However, hyperinsulinemia also promotes lipid uptake, fat synthesis, and fat storage. The net result is hepatic triglyceride accumulation (i.e., steatosis).
Triglyceride per se is not hepatotoxic. However, its precursors (e.g., fatty acids and diacylglycerols) and metabolic by-products (e.g., reactive oxygen species) may damage hepatocytes, leading to hepatocyte lipotoxicity. Lipotoxicity also triggers the generation of other factors (e.g., inflammatory cytokines, hormonal mediators) that deregulate systems that normally maintain hepatocyte viability. The net result is increased hepatocyte death. Dying hepatocytes, in turn, release various factors that trigger wound healing responses that aim to replace (regenerate) lost hepatocytes. Such repair involves transient expansion of other cell types, such as myofibroblasts and progenitor cells, that make and degrade matrix, remodel the vasculature, and generate replacement hepatocytes, as well as the recruitment of immune cells that release factors that modulate liver injury and repair. NASH is the morphologic manifestation of lipotoxicity and resultant wound healing responses. Because the severity and duration of lipotoxic liver injury dictate the intensity and duration of repair, the histologic features and outcomes of NASH are variable. Cirrhosis and liver cancer are potential outcomes of chronic NASH. Cirrhosis results from futile repair, i.e., progressive accumulation of wound healing cells, fibrous matrix, and abnormal vasculature (scarring), rather than efficient reconstruction/regeneration of healthy hepatic parenchyma. Primary liver cancers develop when malignantly transformed liver cells escape mechanisms that normally control regenerative growth. The mechanisms responsible for futile repair (cirrhosis) and liver carcinogenesis are not well understood. Because normal liver regeneration is a very complex process, there are multiple opportunities for deregulation and, thus, pathogenic heterogeneity. To date, this heterogeneity has confounded development of both diagnostic tests and treatments for defective/deregulated liver repair (i.e., cirrhosis and cancer). Hence, current strategies focus on circumventing misrepair by preventing and/or reducing lipotoxic liver injury.
DIAGNOSIS
Diagnosing NAFLD requires demonstration of increased liver fat in the absence of hazardous levels of alcohol consumption. Thresholds for potentially dangerous alcohol ingestion have been set at more than one drink per day in women and two drinks per day in men based on epidemiologic evidence that the prevalence of serum aminotransferase elevations increases when alcohol consumption habitually exceeds these levels. In those studies, one drink was defined as having 10 g of ethanol and, thus, is equivalent to one can of beer, 4 ounces of wine, or 1.5 ounces (one shot) of distilled spirits. Other causes of liver fat accumulation (particularly exposure to certain drugs; Table 364-2) and liver injury (e.g., viral hepatitis, autoimmune liver disease, iron or copper overload, α1 antitrypsin deficiency) must also be excluded. Thus, establishing the diagnosis of NAFLD does not require invasive testing: it can be accomplished by history and physical examination, liver imaging (ultrasound is an acceptable first-line test; computed tomography [CT] or magnetic resonance imaging [MRI] enhances sensitivity for liver fat detection but adds expense), and blood tests to exclude other liver diseases. It is important to emphasize that the liver may not be enlarged, and serum aminotransferases and liver function tests (e.g., bilirubin, albumin, prothrombin time) may be completely normal, in individuals with NAFLD. Because there is yet no one specific blood test for NAFLD, confidence in the diagnosis of NAFLD is increased by identification of NAFLD risk factors. The latter include increased body mass index, insulin resistance/type 2 diabetes mellitus, and other parameters indicative of the metabolic syndrome (e.g., systemic hypertension, dyslipidemia, hyperuricemia/gout, cardiovascular disease; Chap. 422) in the patient or family members.
MEDICATIONS ASSOCIATED WITH HEPATIC STEATOSIS |
Establishing the severity of NAFLD-related liver injury and related scarring (i.e., staging NAFLD) is more difficult than simply diagnosing NAFLD. Staging is critically important, however, because it is necessary to define prognosis and thereby determine treatment recommendations. The goal of staging is to distinguish patients with NASH from those with simple steatosis and to identify which of the NASH patients have advanced fibrosis. The 10-year probability of developing liver-related morbidity or mortality in steatosis is negligible, and hence, this subgroup of NAFLD patients tends to be managed conservatively (see below). In contrast, more intensive follow-up and therapy are justified in NASH patients, and the subgroup with advanced fibrosis merits the most intensive scrutiny and intervention because their 10-year risk of liver-related morbidity and mortality is clearly increased.
Staging approaches can be separated into noninvasive testing (i.e., blood testing, physical examination, and imaging) and invasive approaches (i.e., liver biopsy). Blood test evidence of hepatic dysfunction (e.g., hyperbilirubinemia, hypoalbuminemia, prothrombin time prolongation) or portal hypertension (e.g., thrombocytopenia) and stigmata of portal hypertension on physical examination (e.g., spider angiomata, palmar erythema, splenomegaly, ascites, clubbing, encephalopathy) suggest a diagnosis of advanced NAFLD. Currently, however, liver biopsy is the gold standard for establishing the severity of liver injury and fibrosis because it is both more sensitive and specific than these other tests for establishing NAFLD severity. Although invasive, liver biopsy is seldom complicated by serious adverse sequelae such as significant bleeding, pain, or inadvertent puncture of other organs and thus is relatively safe. However, biopsy suffers from potential sampling error unless tissue cores of 2 cm or longer are acquired. Also, examination of tissue at a single point in time is not reliable for determining whether the pathologic processes are progressing or regressing. The risk of serial liver biopsies within short time intervals is generally deemed as unacceptable outside of research studies. These limitations of liver biopsy have stimulated efforts to develop noninvasive approaches to stage NAFLD. As is true for many other types of chronic liver disease, in NAFLD the levels of serum aminotransferases (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) do not reliably reflect the severity of liver cell injury, extent of liver cell death, or related liver inflammation and fibrosis. Thus, they are imperfect for determining which individuals with NAFLD have NASH. This has stimulated research to identify superior markers of liver injury. Serum levels of keratin 8 and keratin 18 appear to be promising surrogates. Keratins 8 and 18 (K8/18) are epithelial cytoskeletal proteins that undergo cleavage during programmed cell death (apoptosis). Both cleaved and full-length K8/18 are released into the blood as hepatocytes die, and studies suggest that serum levels of K8/18 differentiate individuals with NASH from those with simple steatosis or normal livers more reliably than do serum aminotransferase levels. Moreover, K8/18 levels appear to parallel the severity of liver fibrosis, with higher levels marking individuals who are likely to have worse scarring (i.e., advanced liver fibrosis or cirrhosis). While promising, testing for K8/18 has not yet become standard clinical practice. Other blood tests and imaging approaches that quantify liver fibrosis are also being developed. Recently, the U.S. Food and Drug Administration (FDA) approved an ultrasound-based test that measures liver stiffness as a surrogate marker of fibrosis (FibroScan®) (Chap. 358). This new tool will likely be used serially to monitor fibrosis progression and regression in NAFLD patients. Studies that compare the receiver operator characteristics of K8/18 plus FibroScan® versus liver biopsy for monitoring NAFLD evolution are forthcoming.
CLINICAL FEATURES OF NAFLD
Most subjects with NAFLD are asymptomatic. The diagnosis is often made when abnormal liver aminotransferases or features of fatty liver are noted during an evaluation performed for other reasons. NAFLD may also be diagnosed during the workup of vague right upper quadrant abdominal pain, hepatomegaly, or an abnormal-appearing liver at time of abdominal surgery. Obesity is present in 50–90% of subjects. Most patients with NAFLD also have other features of the metabolic syndrome (Chap. 422). Some have subtle stigmata of chronic liver disease, such as spider angiomata, palmer erythema, or splenomegaly. In a small minority of patients with advanced NAFLD, complications of end-stage liver disease (e.g., jaundice, features of portal hypertension such as ascites or variceal hemorrhage) may be the initial findings.
The association of NAFLD with obesity, diabetes, hypertriglyceridemia, hypertension, and cardiovascular disease is well known. Other associations include chronic fatigue, mood alterations, obstructive sleep apnea, thyroid dysfunction, and chronic pain syndrome. NAFLD is an independent risk factor for metabolic syndrome (Chap. 422). Longitudinal studies suggest that patients with NASH are at two- to threefold increased risk for the development of metabolic syndrome. Similarly, studies have shown that patients with NASH have a higher risk for the development of hypertension and diabetes mellitus. The presence of NAFLD is also independently associated with endothelial dysfunction, increased carotid intimal thickness, and the number of plaques in carotid and coronary arteries. Such data indicate that NAFLD has many deleterious effects on health in general.
TREATMENT OF NAFLD
Treatment of NAFLD can be divided into three components: (1) specific therapy of NAFLD-related liver disease; (2) treatment of NAFLD-associated comorbidities; and (3) treatment of the complications of advanced NAFLD. The subsequent discussion focuses on specific therapies for NAFLD, with some mention of their impact on major NAFLD comorbidities (insulin resistance/diabetes, obesity, and dyslipidemia). Treatment of the complications of advanced NAFLD involves management of the complications of cirrhosis and portal hypertension, including primary liver cancers. Approaches to accomplish these objectives are similar to those used in other chronic liver diseases and are covered elsewhere in the textbook (Chaps. 365 and 111).
At present, there are no FDA-approved therapies for the treatment of NAFLD. Thus, the current approach to NAFLD management focuses on treatment to improve the risk factors for NASH (i.e., obesity, insulin resistance, metabolic syndrome, dyslipidemia). Based on our understanding of the natural history of NAFLD, only patients with NASH or those with features of hepatic fibrosis on liver biopsy are considered currently for targeted pharmacologic therapies. This approach may change as our understanding of disease pathophysiology improves and potential targets of therapy evolve.
Diet and Exercise Lifestyle changes and dietary modification are the foundation for NAFLD treatment. Many studies indicate that lifestyle modification can improve serum aminotransferases and hepatic steatosis, with loss of at least 3–5% of body weight improving steatosis, but greater weight loss (up to 10%) necessary to improve steatohepatitis. The benefits of different dietary macronutrient contents (e.g., low-carbohydrate vs low-fat diets, saturated vs unsaturated fat diets) and different intensities of calorie restriction appear to be comparable. In adults with NAFLD, exercise regimens that improve fitness may be sufficient to reduce hepatic steatosis, but their impact on other aspects of liver histology remains unknown. Unfortunately, most NAFLD patients are unable to achieve sustained weight loss. Although pharmacologic therapies such as orlistat, topiramate, and phentermine to facilitate weight loss are available, their role in the treatment of NAFLD remains experimental.
Pharmacologic Therapies Several drug therapies have been tried in both research and clinical settings. No agent has yet been approved by the FDA for the treatment of NAFLD. Hence, this remains an area of active research. Because NAFLD is strongly associated with the metabolic syndrome and type 2 diabetes (Chaps. 417 and 418), the efficacy of various insulin-sensitizing agents has been examined. Metformin, an agent that mainly improves hepatic insulin sensitivity, has been evaluated in several small, open-label studies in adults and a recent larger, prospectively randomized trial in children (dubbed the TONIC study). Although several of the adult NASH studies suggested improvements in aminotransferases and/or liver histology, metformin did not improve liver histology in the TONIC study of children with NASH. Thus, it is not currently recommended as a treatment for NASH. Uncontrolled open-label studies have also investigated thiazolidinediones (pioglitazone and rosiglitazone) in adults with NASH. This class of drugs is known to improve systemic insulin resistance. Both pioglitazone and rosiglitazone reduced aminotransferases and improved some of the histologic features of NASH in small, uncontrolled studies. A large, National Institutes of Health–sponsored, randomized placebo-controlled clinical trial, the PIVENs Study (Pioglitazone vs Vitamin E vs Placebo for the Treatment of 247 Nondiabetic Adults with NASH), demonstrated that resolution of histologic NASH occurred more often in subjects treated with pioglitazone (30 mg/d) than with placebo for 18 months (47 vs 21%, p = .001). However, many subjects in the pioglitazone group gained weight, and liver fibrosis did not improve. Also, it should be noted that the long-term safety and efficacy of thiazolidinediones in patients with NASH has not been established. Five-year follow-up of subjects treated with rosiglitazone demonstrated no reduction in liver fibrosis, and rosiglitazone has been associated with increased long-term risk for cardiovascular mortality. Hence, it is not recommended as a treatment for NAFLD. Pioglitazone may be safer because in a recent large meta-analysis it was associated with reduced overall morality, myocardial infarction, and stroke. However, caution must be exercised when considering its use in patients with impaired myocardial function.
Antioxidants have also been evaluated for the treatment of NAFLD because oxidant stress is thought to contribute to the pathogenesis of NASH. Vitamin E, an inexpensive yet potent antioxidant, has been examined in several small pediatric and adult studies with varying results. In all of those studies, vitamin E was well tolerated, and most showed modest improvements in aminotransferase levels, radiographic features of hepatic steatosis, and/or histologic features of NASH. Vitamin E (800 IU/d) was also compared to placebo in the PIVENs and TONIC studies. In PIVENs, vitamin E was the only agent that achieved the predetermined primary endpoint (i.e., improvement in steatohepatitis, lobular inflammation, and steatosis score, without an increase in the fibrosis score). This endpoint was met in 43% of patients in the vitamin E group (p = .001 vs placebo), 34% in the pioglitazone group (p = .04 vs placebo), and 19% in the placebo group. Vitamin E also improved NASH histology in pediatric patients with NASH (TONIC trial). However, a recent population-based study suggested that chronic vitamin E therapy may increase the risk for cardiovascular mortality. Thus, vitamin E should only be considered as a first-line pharmacotherapy for nondiabetic NASH patients. Also, given its potentially negative effects on cardiovascular health, caution should be exercised until the risk-to-benefit ratio and long-term therapeutic efficacy of vitamin E are better defined. Ursodeoxycholic acid (a bile acid that improves certain cholestatic liver diseases) and betaine (metabolite of choline that raises SAM levels and decreases cellular oxidative damage) offer no histologic benefit over placebo in patients with NASH. Experimental evidence to support the use of omega-3 fatty acids in NAFLD exists; however, a recent large, multicenter, placebo-controlled study failed to demonstrate a histologic benefit. Other pharmacotherapies are also being evaluated in NAFLD (e.g., probiotics, farnesoid × receptor agonists, anticytokine agents, glucagon-like peptide agonists, dipeptidyl IV antagonists); however, sufficient data do not yet exist to justify their use as NASH treatments in standard clinical practice.
Statins are an important class of agents to treat dyslipidemia and decrease cardiovascular risk. There is no evidence to suggest that statins cause liver failure in patients with any chronic liver disease, including NAFLD. The incidence of liver enzyme elevations in NAFLD patients taking statins is also no different than that of healthy controls or patients with other chronic liver diseases. Moreover, several studies have suggested that statins may improve aminotransferases and histology in patients with NASH. Yet, there is continued reluctance to use statins in patients with NAFLD. The lack of evidence that statins harm the liver in NAFLD patients, combined with the increase risk for cardiovascular morbidity and mortality in NAFLD patients, warrants the use of statins to treat dyslipidemia in patients with NAFLD/NASH.
Bariatric Surgery Although interest in bariatric surgery as a treatment for NAFLD exists, a recently published Cochrane review concluded that lack of randomized clinical trials or adequate clinical studies prevents definitive assessment of benefits and harms of bariatric surgery as a treatment for NASH. Most studies of bariatric surgery have shown that bariatric surgery is generally safe in individuals with well-compensated chronic liver disease and improves hepatic steatosis and necroinflammation (i.e., features of NAFLD/NASH); however, effects on hepatic fibrosis have been variable. Concern lingers because some of the largest prospective studies suggest that hepatic fibrosis might progress after bariatric surgery. Thus, the Cochrane review deemed it premature to recommend bariatric surgery as a primary treatment for NASH. There is also general agreement that patients with NAFLD-related cirrhosis and portal hypertension should be excluded as candidates for bariatric surgery. However, given growing evidence for the benefits of bariatric surgery on metabolic syndrome complications in individuals with refractory obesity, it is not contraindicated in otherwise eligible patients with NAFLD or NASH.
Liver Transplantation Patients with NAFLD in whom end-stage liver disease develops should be evaluated for liver transplantation (Chap. 368). The outcomes of liver transplantation in well-selected patients with NAFLD are generally good, but comorbid medical conditions associated with NAFLD, such as diabetes mellitus, obesity, and cardiovascular disease, often limit transplant candidacy. NAFLD may recur after liver transplantation. The risk factors for recurrent or de novo NAFLD after liver transplantation are multifactorial and include hypertriglyceridemia, obesity, diabetes mellitus, and immunosuppressive therapies, particularly glucocorticoids.
GLOBAL HEALTH CONSIDERATIONS
![]() The epidemic of obesity is now a global and accelerating phenomenon. Worldwide, there are over 1 billion overweight adults, of whom at least 300 million are obese. In the wake of the obesity epidemic follow numerous comorbidities, including NAFLD. NAFLD is the most common liver disease identified in Western countries and the fastest rising form of chronic liver disease worldwide. Present understanding of NAFLD natural history is based mainly on studies in whites who became overweight/obese and developed the metabolic syndrome in adulthood. The impact of the global childhood obesity epidemic on NAFLD pathogenesis/progression is unknown. Emerging evidence demonstrates that advanced NAFLD, including cirrhosis and primary liver cancer, can occur in children, prompting concerns that childhood-onset NAFLD might follow a more aggressive course than typical adult-acquired NAFLD. Some of the most populated parts of the world are in the midst of industrial revolutions, and certain environmental pollutants seem to exacerbate NAFLD. Some studies also suggest that the risk for NASH and NAFLD-related cirrhosis may be higher in certain ethnic groups such as Asians, certain Hispanics, and Native Americans and lower in others such as African Americans, compared with whites. Although all of these variables confound efforts to predict the net impact of this obesity-related liver disease on global health, it seems likely that NAFLD will remain a major cause of chronic liver disease worldwide for the foreseeable future.
The epidemic of obesity is now a global and accelerating phenomenon. Worldwide, there are over 1 billion overweight adults, of whom at least 300 million are obese. In the wake of the obesity epidemic follow numerous comorbidities, including NAFLD. NAFLD is the most common liver disease identified in Western countries and the fastest rising form of chronic liver disease worldwide. Present understanding of NAFLD natural history is based mainly on studies in whites who became overweight/obese and developed the metabolic syndrome in adulthood. The impact of the global childhood obesity epidemic on NAFLD pathogenesis/progression is unknown. Emerging evidence demonstrates that advanced NAFLD, including cirrhosis and primary liver cancer, can occur in children, prompting concerns that childhood-onset NAFLD might follow a more aggressive course than typical adult-acquired NAFLD. Some of the most populated parts of the world are in the midst of industrial revolutions, and certain environmental pollutants seem to exacerbate NAFLD. Some studies also suggest that the risk for NASH and NAFLD-related cirrhosis may be higher in certain ethnic groups such as Asians, certain Hispanics, and Native Americans and lower in others such as African Americans, compared with whites. Although all of these variables confound efforts to predict the net impact of this obesity-related liver disease on global health, it seems likely that NAFLD will remain a major cause of chronic liver disease worldwide for the foreseeable future.
365 | Cirrhosis and Its Complications |
Cirrhosis is a condition that is defined histopathologically and has a variety of clinical manifestations and complications, some of which can be life-threatening. In the past, it has been thought that cirrhosis was never reversible; however, it has become apparent that when the underlying insult that has caused the cirrhosis has been removed, there can be reversal of fibrosis. This is most apparent with the successful treatment of chronic hepatitis C; however, reversal of fibrosis is also seen in patients with hemochromatosis who have been successfully treated and in patients with alcoholic liver disease who have discontinued alcohol use.
Regardless of the cause of cirrhosis, the pathologic features consist of the development of fibrosis to the point that there is architectural distortion with the formation of regenerative nodules. This results in a decrease in hepatocellular mass, and thus function, and an alteration of blood flow. The induction of fibrosis occurs with activation of hepatic stellate cells, resulting in the formation of increased amounts of collagen and other components of the extracellular matrix.
Clinical features of cirrhosis are the result of pathologic changes and mirror the severity of the liver disease. Most hepatic pathologists provide an assessment of grading and staging when evaluating liver biopsy samples. These grading and staging schemes vary between disease states and have been developed for most conditions, including chronic viral hepatitis, nonalcoholic fatty liver disease, and primary biliary cirrhosis. Advanced fibrosis usually includes bridging fibrosis with nodularity designated as stage 3 and cirrhosis designated as stage 4. Patients who have cirrhosis have varying degrees of compensated liver function, and clinicians need to differentiate between those who have stable, compensated cirrhosis and those who have decompensated cirrhosis. Patients who have developed complications of their liver disease and have become decompensated should be considered for liver transplantation. Many of the complications of cirrhosis will require specific therapy. Portal hypertension is a significant complicating feature of decompensated cirrhosis and is responsible for the development of ascites and bleeding from esophagogastric varices, two complications that signify decompensated cirrhosis. Loss of hepatocellular function results in jaundice, coagulation disorders, and hypoalbuminemia and contributes to the causes of portosystemic encephalopathy. The complications of cirrhosis are basically the same regardless of the etiology. Nonetheless, it is useful to classify patients by the cause of their liver disease (Table 365-1); patients can be divided into broad groups with alcoholic cirrhosis, cirrhosis due to chronic viral hepatitis, biliary cirrhosis, and other, less common causes such as cardiac cirrhosis, cryptogenic cirrhosis, and other miscellaneous causes.
CAUSES OF CIRRHOSIS |
ALCOHOLIC CIRRHOSIS
Excessive chronic alcohol use can cause several different types of chronic liver disease, including alcoholic fatty liver, alcoholic hepatitis, and alcoholic cirrhosis. Furthermore, use of excessive alcohol can contribute to liver damage in patients with other liver diseases, such as hepatitis C, hemochromatosis, and fatty liver disease related to obesity. Chronic alcohol use can produce fibrosis in the absence of accompanying inflammation and/or necrosis. Fibrosis can be centrilobular, pericellular, or periportal. When fibrosis reaches a certain degree, there is disruption of the normal liver architecture and replacement of liver cells by regenerative nodules. In alcoholic cirrhosis, the nodules are usually <3 mm in diameter; this form of cirrhosis is referred to as micronodular. With cessation of alcohol use, larger nodules may form, resulting in a mixed micronodular and macronodular cirrhosis.
Pathogenesis Alcohol is the most commonly used drug in the United States, and more than two-thirds of adults drink alcohol each year. Thirty percent have had a binge within the past month, and over 7% of adults regularly consume more than two drinks per day. Unfortunately, more than 14 million adults in the United States meet the diagnostic criteria for alcohol abuse or dependence. In the United States, chronic liver disease is the tenth most common cause of death in adults, and alcoholic cirrhosis accounts for approximately 40% of deaths due to cirrhosis.
Ethanol is mainly absorbed by the small intestine and, to a lesser degree, through the stomach. Gastric alcohol dehydrogenase (ADH) initiates alcohol metabolism. Three enzyme systems account for metabolism of alcohol in the liver. These include cytosolic ADH, the microsomal ethanol oxidizing system (MEOS), and peroxisomal catalase. The majority of ethanol oxidation occurs via ADH to form acetaldehyde, which is a highly reactive molecule that may have multiple effects. Ultimately, acetaldehyde is metabolized to acetate by aldehyde dehydrogenase (ALDH). Intake of ethanol increases intracellular accumulation of triglycerides by increasing fatty acid uptake and by reducing fatty acid oxidation and lipoprotein secretion. Protein synthesis, glycosylation, and secretion are impaired. Oxidative damage to hepatocyte membranes occurs due to the formation of reactive oxygen species; acetaldehyde is a highly reactive molecule that combines with proteins to form protein-acetaldehyde adducts. These adducts may interfere with specific enzyme activities, including microtubular formation and hepatic protein trafficking. With acetaldehyde-mediated hepatocyte damage, certain reactive oxygen species can result in Kupffer cell activation. As a result, profibrogenic cytokines are produced that initiate and perpetuate stellate cell activation, with the resultant production of excess collagen and extracellular matrix. Connective tissue appears in both periportal and pericentral zones and eventually connects portal triads with central veins forming regenerative nodules. Hepatocyte loss occurs, and with increased collagen production and deposition, together with continuing hepatocyte destruction, the liver contracts and shrinks in size. This process generally takes from years to decades to occur and requires repeated insults.
Clinical Features The diagnosis of alcoholic liver disease requires an accurate history regarding both amount and duration of alcohol consumption. Patients with alcoholic liver disease can present with nonspecific symptoms such as vague right upper quadrant abdominal pain, fever, nausea and vomiting, diarrhea, anorexia, and malaise. Alternatively, they may present with more specific complications of chronic liver disease, including ascites, edema, or upper gastrointestinal (GI) hemorrhage. Many cases present incidentally at the time of autopsy or elective surgery. Other clinical manifestations include the development of jaundice or encephalopathy. The abrupt onset of any of these complications may be the first event prompting the patient to seek medical attention. Other patients may be identified in the course of an evaluation of routine laboratory studies that are found to be abnormal. On physical examination, the liver and spleen may be enlarged, with the liver edge being firm and nodular. Other frequent findings include scleral icterus, palmar erythema (Fig. 365-1), spider angiomas (Fig. 365-2), parotid gland enlargement, digital clubbing, muscle wasting, or the development of edema and ascites. Men may have decreased body hair and gynecomastia as well as testicular atrophy, which may be a consequence of hormonal abnormalities or a direct toxic effect of alcohol on the testes. In women with advanced alcoholic cirrhosis, menstrual irregularities usually occur, and some women may be amenorrheic. These changes are often reversible following cessation of alcohol.
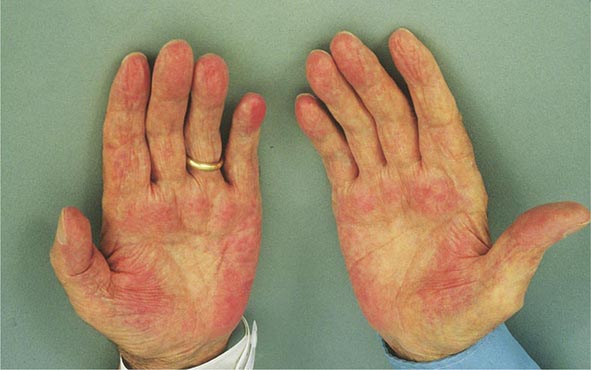
FIGURE 365-1 Palmar erythema. This figure shows palmar erythema in a patient with alcoholic cirrhosis. The erythema is peripheral over the palm with central pallor.
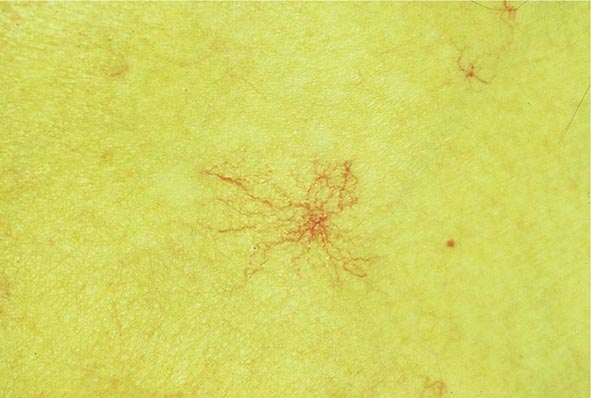
FIGURE 365-2 Spider angioma. This figure shows a spider angioma in a patient with hepatitis C cirrhosis. With release of central compression, the arteriole fills from the center and spreads out peripherally.
Laboratory tests may be completely normal in patients with early compensated alcoholic cirrhosis. Alternatively, in advanced liver disease, many abnormalities usually are present. Patients may be anemic either from chronic GI blood loss, nutritional deficiencies, or hypersplenism related to portal hypertension, or as a direct suppressive effect of alcohol on the bone marrow. A unique form of hemolytic anemia (with spur cells and acanthocytes) called Zieve’s syndrome can occur in patients with severe alcoholic hepatitis. Platelet counts are often reduced early in the disease, reflective of portal hypertension with hypersplenism. Serum total bilirubin can be normal or elevated with advanced disease. Direct bilirubin is frequently mildly elevated in patients with a normal total bilirubin, but the abnormality typically progresses as the disease worsens. Prothrombin times are often prolonged and usually do not respond to administration of parenteral vitamin K. Serum sodium levels are usually normal unless patients have ascites and then can be depressed, largely due to ingestion of excess free water. Serum alanine and aspartate aminotransferases (ALT, AST) are typically elevated, particularly in patients who continue to drink, with AST levels being higher than ALT levels, usually by a 2:1 ratio.
Diagnosis Patients who have any of the above-mentioned clinical features, physical examination findings, or laboratory studies should be considered to have alcoholic liver disease. The diagnosis, however, requires accurate knowledge that the patient is continuing to use and abuse alcohol. Furthermore, other forms of chronic liver disease (e.g., chronic viral hepatitis or metabolic or autoimmune liver diseases) must be considered or ruled out, or if present, an estimate of relative causality along with the alcohol use should be determined. Liver biopsy can be helpful to confirm a diagnosis, but generally when patients present with alcoholic hepatitis and are still drinking, liver biopsy is withheld until abstinence has been maintained for at least 6 months to determine residual, nonreversible disease.
In patients who have had complications of cirrhosis and who continue to drink, there is a <50% 5-year survival. In contrast, in patients who are able to remain abstinent, the prognosis is significantly improved. In patients with advanced liver disease, the prognosis remains poor; however, in individuals who are able to remain abstinent, liver transplantation is a viable option.
CIRRHOSIS DUE TO CHRONIC VIRAL HEPATITIS B OR C
![]() Of patients exposed to the hepatitis C virus (HCV), approximately 80% develop chronic hepatitis C, and of those, about 20–30% will develop cirrhosis over 20–30 years. Many of these patients have had concomitant alcohol use, and the true incidence of cirrhosis due to hepatitis C alone is unknown. Nonetheless, this represents a significant number of patients. It is expected that an even higher percentage will go on to develop cirrhosis over longer periods of time. In the United States, approximately 5 to 6 million people have been exposed to HCV, with about 4 million who are chronically viremic. Worldwide, about 170 million individuals have hepatitis C, with some areas of the world (e.g., Egypt) having up to 15% of the population infected. HCV is a noncytopathic virus, and liver damage is probably immune-mediated. Progression of liver disease due to chronic hepatitis C is characterized by portal-based fibrosis with bridging fibrosis and nodularity developing, ultimately culminating in the development of cirrhosis. In cirrhosis due to chronic hepatitis C, the liver is small and shrunken with characteristic features of a mixed micro- and macronodular cirrhosis seen on liver biopsy. In addition to the increased fibrosis that is seen in cirrhosis due to hepatitis C, an inflammatory infiltrate is found in portal areas with interface hepatitis and occasionally some lobular hepatocellular injury and inflammation. In patients with HCV genotype 3, steatosis is often present.
Of patients exposed to the hepatitis C virus (HCV), approximately 80% develop chronic hepatitis C, and of those, about 20–30% will develop cirrhosis over 20–30 years. Many of these patients have had concomitant alcohol use, and the true incidence of cirrhosis due to hepatitis C alone is unknown. Nonetheless, this represents a significant number of patients. It is expected that an even higher percentage will go on to develop cirrhosis over longer periods of time. In the United States, approximately 5 to 6 million people have been exposed to HCV, with about 4 million who are chronically viremic. Worldwide, about 170 million individuals have hepatitis C, with some areas of the world (e.g., Egypt) having up to 15% of the population infected. HCV is a noncytopathic virus, and liver damage is probably immune-mediated. Progression of liver disease due to chronic hepatitis C is characterized by portal-based fibrosis with bridging fibrosis and nodularity developing, ultimately culminating in the development of cirrhosis. In cirrhosis due to chronic hepatitis C, the liver is small and shrunken with characteristic features of a mixed micro- and macronodular cirrhosis seen on liver biopsy. In addition to the increased fibrosis that is seen in cirrhosis due to hepatitis C, an inflammatory infiltrate is found in portal areas with interface hepatitis and occasionally some lobular hepatocellular injury and inflammation. In patients with HCV genotype 3, steatosis is often present.
![]() Similar findings are seen in patients with cirrhosis due to chronic hepatitis B. Of adult patients exposed to hepatitis B, about 5% develop chronic hepatitis B, and about 20% of those patients will go on to develop cirrhosis. Special stains for hepatitis B core (HBc) and hepatitis B surface (HBs) antigen will be positive, and ground-glass hepatocytes signifying hepatitis B surface antigen (HBsAg) may be present. In the United States, there are about 2 million carriers of hepatitis B, whereas in other parts of the world where hepatitis B virus (HBV) is endemic (i.e., Asia, Southeast Asia, sub-Saharan Africa), up to 15% of the population may be infected, having acquired the infection vertically at the time of birth. Thus, over 300–400 million individuals are thought to have hepatitis B worldwide. Approximately 25% of these individuals may ultimately develop cirrhosis.
Similar findings are seen in patients with cirrhosis due to chronic hepatitis B. Of adult patients exposed to hepatitis B, about 5% develop chronic hepatitis B, and about 20% of those patients will go on to develop cirrhosis. Special stains for hepatitis B core (HBc) and hepatitis B surface (HBs) antigen will be positive, and ground-glass hepatocytes signifying hepatitis B surface antigen (HBsAg) may be present. In the United States, there are about 2 million carriers of hepatitis B, whereas in other parts of the world where hepatitis B virus (HBV) is endemic (i.e., Asia, Southeast Asia, sub-Saharan Africa), up to 15% of the population may be infected, having acquired the infection vertically at the time of birth. Thus, over 300–400 million individuals are thought to have hepatitis B worldwide. Approximately 25% of these individuals may ultimately develop cirrhosis.
Clinical Features and Diagnosis Patients with cirrhosis due to either chronic hepatitis C or B can present with the usual symptoms and signs of chronic liver disease. Fatigue, malaise, vague right upper quadrant pain, and laboratory abnormalities are frequent presenting features. Diagnosis requires a thorough laboratory evaluation, including quantitative HCV RNA testing and analysis for HCV genotype, or hepatitis B serologies to include HBsAg, anti-HBs, HBeAg (hepatitis B e antigen), anti-HBe, and quantitative HBV DNA levels.
CIRRHOSIS FROM AUTOIMMUNE HEPATITIS AND NONALCOHOLIC FATTY LIVER DISEASE
Other causes of posthepatitic cirrhosis include autoimmune hepatitis and cirrhosis due to nonalcoholic steatohepatitis. Many patients with autoimmune hepatitis (AIH) present with cirrhosis that is already established. Typically, these patients will not benefit from immunosuppressive therapy with glucocorticoids or azathioprine because the AIH is “burned out.” In this situation, liver biopsy does not show a significant inflammatory infiltrate. Diagnosis in this setting requires positive autoimmune markers such as antinuclear antibody (ANA) or anti-smooth-muscle antibody (ASMA). When patients with AIH present with cirrhosis and active inflammation accompanied by elevated liver enzymes, there can be considerable benefit from the use of immunosuppressive therapy.
Patients with nonalcoholic steatohepatitis are increasingly being found to have progressed to cirrhosis. With the epidemic of obesity that continues in Western countries, more and more patients are identified with nonalcoholic fatty liver disease (Chap. 364). Of these, a significant subset has nonalcoholic steatohepatitis and can progress to increased fibrosis and cirrhosis. Over the past several years, it has been increasingly recognized that many patients who were thought to have cryptogenic cirrhosis in fact have nonalcoholic steatohepatitis. As their cirrhosis progresses, they become catabolic and then lose the telltale signs of steatosis seen on biopsy. Management of complications of cirrhosis due to either AIH or nonalcoholic steatohepatitis is similar to that for other forms of cirrhosis.
BILIARY CIRRHOSIS
Biliary cirrhosis has pathologic features that are different from either alcoholic cirrhosis or posthepatitic cirrhosis, yet the manifestations of end-stage liver disease are the same. Cholestatic liver disease may result from necroinflammatory lesions, congenital or metabolic processes, or external bile duct compression. Thus, two broad categories reflect the anatomic sites of abnormal bile retention: intrahepatic and extrahepatic. The distinction is important for obvious therapeutic reasons. Extrahepatic obstruction may benefit from surgical or endoscopic biliary tract decompression, whereas intrahepatic cholestatic processes will not improve with such interventions and require a different approach.
The major causes of chronic cholestatic syndromes are primary biliary cirrhosis (PBC), autoimmune cholangitis (AIC), primary sclerosing cholangitis (PSC), and idiopathic adulthood ductopenia. These syndromes are usually clinically distinguished from each other by antibody testing, cholangiographic findings, and clinical presentation. However, they all share the histopathologic features of chronic cholestasis, such as cholate stasis; copper deposition; xanthomatous transformation of hepatocytes; and irregular, so-called biliary fibrosis. In addition, there may be chronic portal inflammation, interface activity, and chronic lobular inflammation. Ductopenia is a result of this progressive disease as patients develop cirrhosis.
PRIMARY BILIARY CIRRHOSIS
PBC is seen in about 100–200 individuals per million, with a strong female preponderance and a median age of around 50 years at the time of diagnosis. The cause of PBC is unknown; it is characterized by portal inflammation and necrosis of cholangiocytes in small- and medium-sized bile ducts. Cholestatic features prevail, and biliary cirrhosis is characterized by an elevated bilirubin level and progressive liver failure. Liver transplantation is the treatment of choice for patients with decompensated cirrhosis due to PBC. A variety of therapies have been proposed, but ursodeoxycholic acid (UDCA) is the only approved treatment that has some degree of efficacy by slowing the rate of progression of the disease.
Antimitochondrial antibodies (AMA) are present in about 90% of patients with PBC. These autoantibodies recognize intermitochondrial membrane proteins that are enzymes of the pyruvate dehydrogenase complex (PDC), the branched-chain 2-oxoacid dehydrogenase complex, and the 2-oxogluterate dehydrogenase complex. Most relate to pyruvate dehydrogenase. These autoantibodies are not pathogenic but rather are useful markers for making a diagnosis of PBC.
Pathology Histopathologic analyses of liver biopsies of patients with PBC have resulted in identifying four distinct stages of the disease as it progresses. The earliest lesion is termed chronic nonsuppurative destructive cholangitis and is a necrotizing inflammatory process of the portal tracts. Medium and small bile ducts are infiltrated with lymphocytes and undergo duct destruction. Mild fibrosis and sometimes bile stasis can occur. With progression, the inflammatory infiltrate becomes less prominent, but the number of bile ducts is reduced and there is proliferation of smaller bile ductules. Increased fibrosis ensues with the expansion of periportal fibrosis to bridging fibrosis. Finally, cirrhosis, which may be micronodular or macronodular, develops.
Clinical Features Currently, most patients with PBC are diagnosed well before the end-stage manifestations of the disease are present, and, as such, most patients are actually asymptomatic. When symptoms are present, they most prominently include a significant degree of fatigue out of proportion to what would be expected for either the severity of the liver disease or the age of the patient. Pruritus is seen in approximately 50% of patients at the time of diagnosis, and it can be debilitating. It might be intermittent and usually is most bothersome in the evening. In some patients, pruritus can develop toward the end of pregnancy, and there are examples of patients having been diagnosed with cholestasis of pregnancy rather than PBC. Pruritus that presents prior to the development of jaundice indicates severe disease and a poor prognosis.
Physical examination can show jaundice and other complications of chronic liver disease, including hepatomegaly, splenomegaly, ascites, and edema. Other features that are unique to PBC include hyperpigmentation, xanthelasma, and xanthomata, which are related to the altered cholesterol metabolism seen in this disease. Hyperpigmentation is evident on the trunk and the arms and is seen in areas of exfoliation and lichenification associated with progressive scratching related to the pruritus. Bone pain resulting from osteopenia or osteoporosis is occasionally seen at the time of diagnosis.
Laboratory Findings Laboratory findings in PBC show cholestatic liver enzyme abnormalities with an elevation in γ-glutamyl transpeptidase and alkaline phosphatase (ALP) along with mild elevations in aminotransferases (ALT and AST). Immunoglobulins, particularly IgM, are typically increased. Hyperbilirubinemia usually is seen once cirrhosis has developed. Thrombocytopenia, leukopenia, and anemia may be seen in patients with portal hypertension and hypersplenism. Liver biopsy shows characteristic features as described above and should be evident to any experienced hepatopathologist. Up to 10% of patients with characteristic PBC will have features of AIH as well and are defined as having “overlap” syndrome. These patients are usually treated as PBC patients and may progress to cirrhosis with the same frequency as typical PBC patients. Some patients require immunosuppressive medications as well.
Diagnosis PBC should be considered in patients with chronic cholestatic liver enzyme abnormalities. It is most often seen in middle-aged women. AMA testing may be negative, and it should be remembered that as many as 10% of patients with PBC may be AMA-negative. Liver biopsy is most important in this setting of AMA-negative PBC. In patients who are AMA-negative with cholestatic liver enzymes, PSC should be ruled out by way of cholangiography.
TREATMENT | PRIMARY BILIARY CIRRHOSIS |
Treatment of the typical manifestations of cirrhosis are no different for PBC than for other forms of cirrhosis. UDCA has been shown to improve both biochemical and histologic features of the disease. Improvement is greatest when therapy is initiated early; the likelihood of significant improvement with UDCA is low in patients with PBC who present with manifestations of cirrhosis. UDCA is given in doses of 13–15 mg/kg per day; the medication is usually well-tolerated, although some patients have worsening pruritus with initiation of therapy. A small proportion of patients may have diarrhea or headache as a side effect of the drug. UDCA has been shown to slow the rate of progression of PBC, but it does not reverse or cure the disease. Patients with PBC require long-term follow-up by a physician experienced with the disease. Certain patients may need to be considered for liver transplantation should their liver disease decompensate.
The main symptoms of PBC are fatigue and pruritus, and symptom management is important. Several therapies have been tried for treatment of fatigue, but none of them have been successful; frequent naps should be encouraged. Pruritus is treated with antihistamines, narcotic receptor antagonists (naltrexone), and rifampin. Cholestyramine, a bile salt–sequestering agent, has been helpful in some patients but is somewhat tedious and difficult to take. Plasmapheresis has been used rarely in patients with severe intractable pruritus. There is an increased incidence of osteopenia and osteoporosis in patients with cholestatic liver disease, and bone density testing should be performed. Treatment with a bisphosphonate should be instituted when bone disease is identified.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


