FIGURE 76e-21 Non-Hodgkin’s lymphoma involving the skin, with typical violaceous, “plum-colored” nodules. (Courtesy of Jean Bolognia, MD; with permission.)
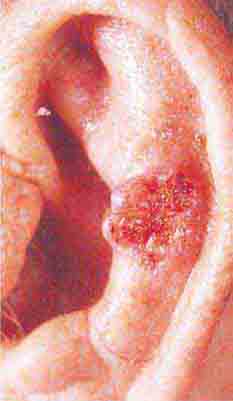
FIGURE 76e-22 Basal cell carcinoma, with central ulceration and a pearly, rolled, telangiectatic tumor border.
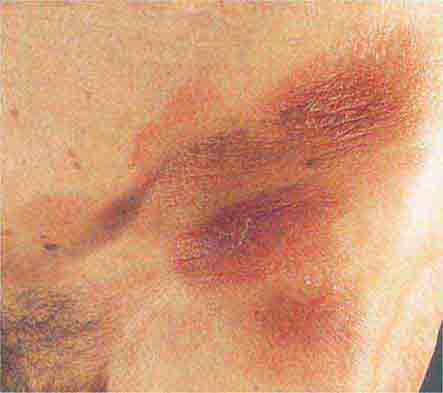
FIGURE 76e-23 Mycosis fungoides is a cutaneous T cell lymphoma. Plaque-stage lesions are seen in this patient.
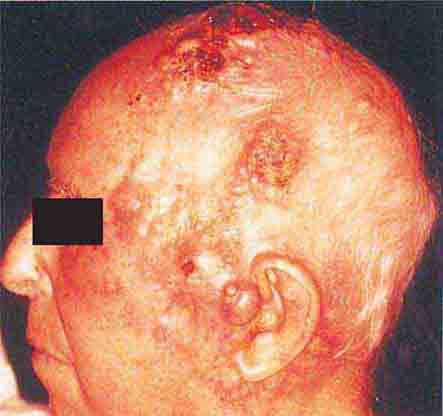
FIGURE 76e-24 Metastatic carcinoma to the skin is characterized by inflammatory, often ulcerated dermal nodules.
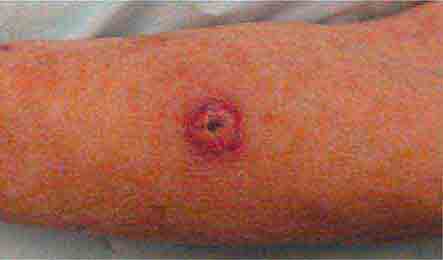
FIGURE 76e-25 Keratoacanthoma is a low-grade squamous cell carcinoma that presents as an exophytic nodule with central keratinous debris.
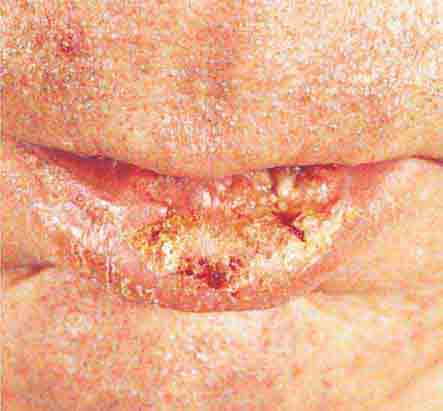
FIGURE 76e-26 Squamous cell carcinoma is seen here as a hyperkeratotic, crusted, and somewhat eroded plaque on the lower lip. Sun-exposed skin of the head, neck, hands, and arms are other typical sites of involvement.
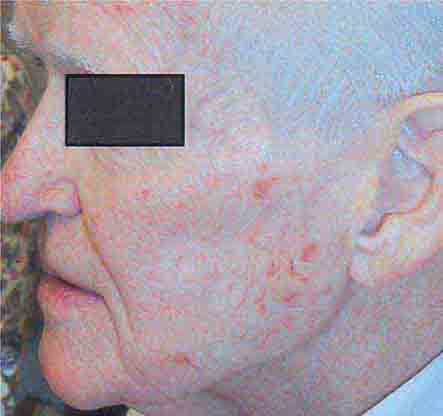
FIGURE 76e-27 Actinic keratoses consist of hyperkeratotic erythematous papules and patches on sun-exposed skin. They arise in middle-aged to older adults and have some potential for malignant transformation. (Courtesy of Robert Swerlick, MD; with permission.)
MELANOMA AND BENIGN PIGMENTED LESIONS
(Figs. 76e-28 to 76e-33) As the prognosis of melanoma is related primarily to the microscopic depth of invasion, and as early detection with surgical treatment can be curative in a high percentage of patients, it is essential that all clinicians acquire some facility in evaluating pigmented lesions. Three clinicopathologic subtypes of melanoma—superficial spreading, lentigo maligna, and acral lentiginous melanoma—typically display features noted in the “ABCD rule”: asymmetry (one half of the lesion varies from the other half); border irregularity (the circumferential border exhibits an irregular, sometimes jagged appearance); color (there is uneven coloration and tone to the pigmented lesion, with various shades of brown, black, red, and white in different areas); and diameter (the diameter is typically >6 mm). The more uncommon subtype, nodular melanoma, may not manifest all these features but rather may present as a more symmetric, evenly pigmented, or amelanotic lesion. Dysplastic (atypical) melanocytic nevi may occur as solitary or multiple lesions as well as in the setting of familial melanoma. These nevi display some degree of asymmetry, border irregularity, and color variation. Ordinary nevi may be acquired or congenital and are quite common.
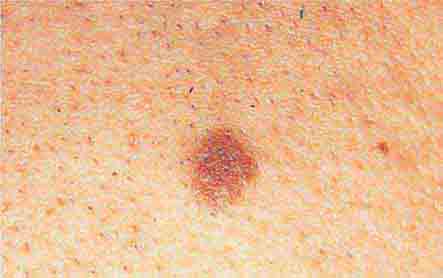
FIGURE 76e-28 Nevi are benign proliferations of nevomelanocytes characterized by regularly shaped hyperpigmented macules or papules of a uniform color.
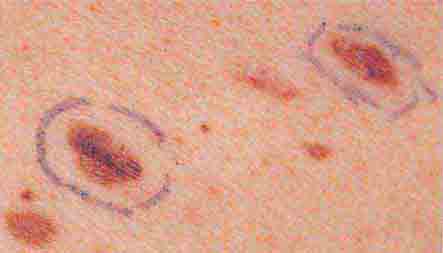
FIGURE 76e-29 Dysplastic nevi are irregularly pigmented and shaped nevomelanocytic lesions that may be associated with familial melanoma.
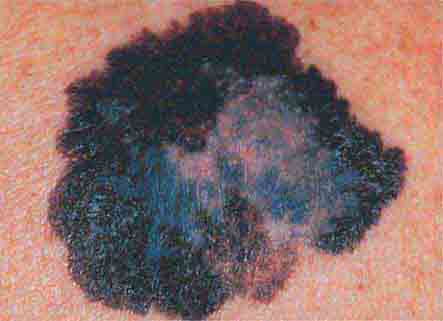
FIGURE 76e-30 Superficial spreading melanoma, the most common type of malignant melanoma, is characterized by color variegation (black, blue, brown, pink, and white) and irregular borders.
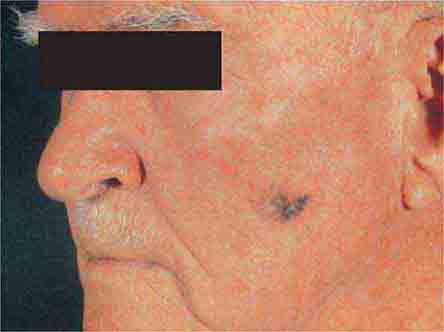
FIGURE 76e-31 Lentigo maligna melanoma occurs on sun-exposed skin as a large, hyperpigmented macule or plaque with irregular borders and variable pigmentation. (Courtesy of Alvin Solomon, MD; with permission.)

FIGURE 76e-32 Nodular melanoma most commonly manifests as a rapidly growing, often ulcerated or crusted black nodule. (Courtesy of S. Wright Caughman, MD; with permission.)
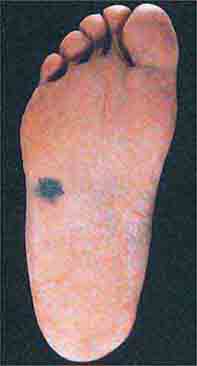
FIGURE 76e-33 Acral lentiginous melanoma is more common among blacks, Asians, and Hispanics and occurs as an enlarging hyperpigmented macule or plaque on the palms or soles. Lateral pigment diffusion is present.
INFECTIOUS DISEASE AND THE SKIN
(Figs. 76e-34 to 76e-58) One of the roles of the skin is to function as a barrier from the outside world. In this capacity, exposure to infectious agents occurs, and bacterial, viral, fungal, and parasitic infections may result. In addition, the skin may be secondarily involved and provides diagnostic clues to systemic infections such as meningococcemia, Rocky Mountain spotted fever, Lyme disease, and septic emboli. Most sexually transmitted bacterial and viral diseases exhibit cutaneous involvement; examples include primary and secondary syphilis, chancroid, genital herpes simplex, and condyloma acuminatum.
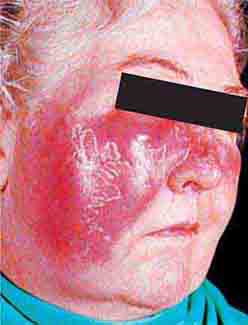
FIGURE 76e-34 Erysipelas is a streptococcal infection of the superficial dermis and consists of well-demarcated, erythematous, edematous, warm plaques.
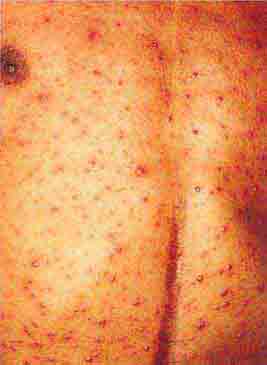
FIGURE 76e-35 Varicella, with numerous lesions in various stages of evolution: vesicles on an erythematous base, umbilicated vesicles, and crusts. (Courtesy of Robert Hartman, MD; with permission.)
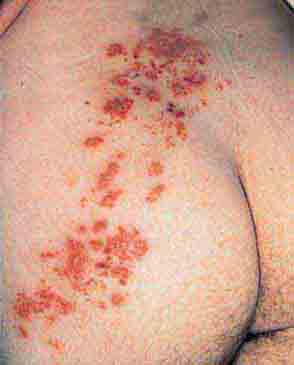
FIGURE 76e-36 Herpes zoster is seen in this HIV-infected patient as hemorrhagic vesicles and pustules on an erythematous base in a dermatomal distribution. (Courtesy of Robert Swerlick, MD; with permission.)
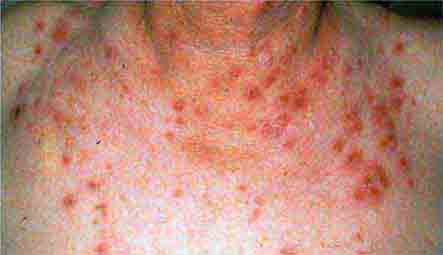
FIGURE 76e-37 Impetigo contagiosa is a superficial streptococcal or Staphylococcus aureus infection consisting of honey-colored crusts and erythematous weeping erosions. Bullous lesions are occasionally seen.
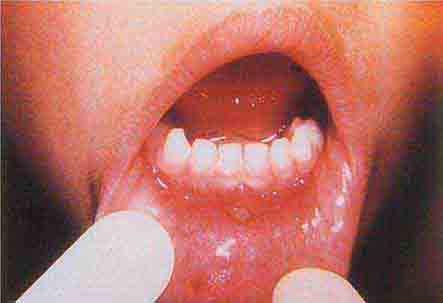
FIGURE 76e-38 Tender vesicles and erosions in the mouth of a patient with hand-foot-and-mouth disease. (Courtesy of Stephen D. Gellis, MD; with permission.)
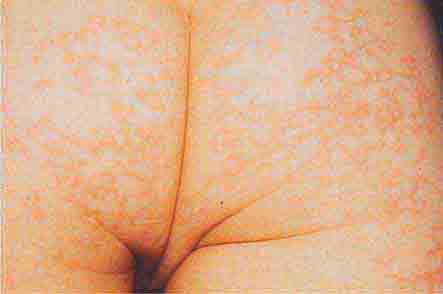
FIGURE 76e-39 Lacy reticular rash of erythema infectiosum (fifth disease).
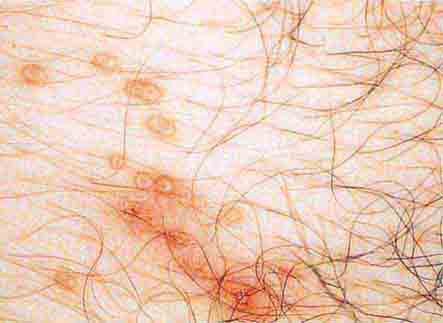
FIGURE 76e-40 Molluscum contagiosum is a cutaneous poxvirus infection characterized by multiple umbilicated flesh-colored or hypopigmented papules. (Courtesy of Yale Resident’s Slide Collection; with permission.)
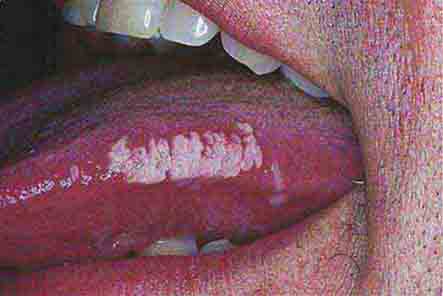
FIGURE 76e-41 Oral hairy leukoplakia often presents as white plaques on the lateral tongue and is associated with Epstein-Barr virus infection. (From K Wolff et al: Fitzpatrick’s Color Atlas & Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005. www.accessmedicine.com.)
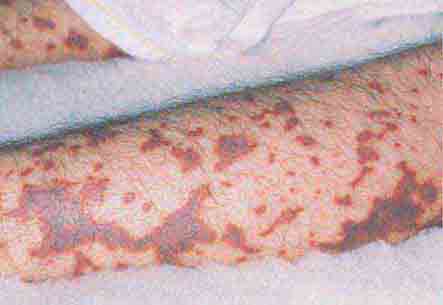
FIGURE 76e-42 Fulminant meningococcemia, with extensive angular purpuric patches. (Courtesy of Stephen D. Gellis, MD; with permission.)
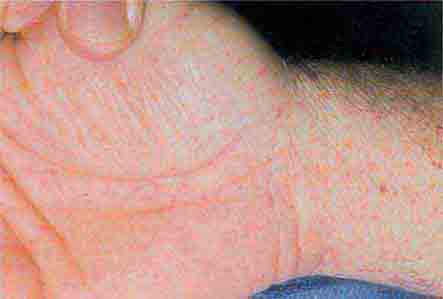
FIGURE 76e-43 Rocky Mountain spotted fever, with pinpoint petechial lesions on the palm and volar aspect of the wrist. (Courtesy of Robert Swerlick, MD; with permission.)
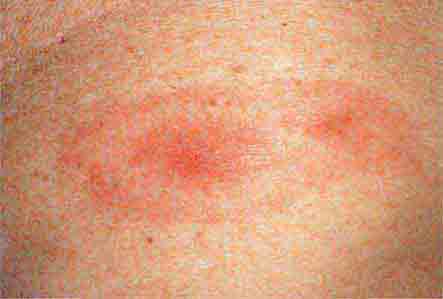
FIGURE 76e-44 Erythema migrans, the early cutaneous manifestation of Lyme disease, is characterized by erythematous annular patches, often with a central erythematous papule at the tick-bite site. (Courtesy of Yale Resident’s Slide Collection; with permission.)
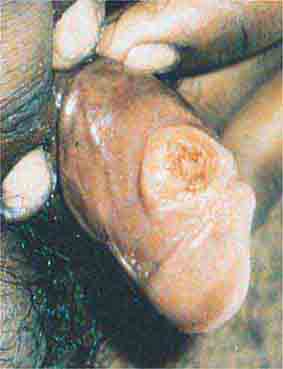
FIGURE 76e-45 Primary syphilis, with a firm, nontender chancre. (Courtesy of Gregory Cox, MD; with permission.)
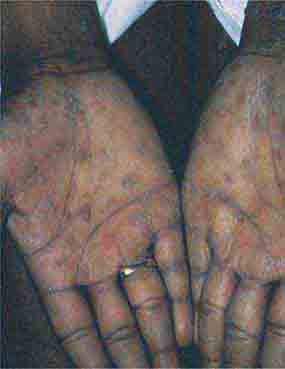
FIGURE 76e-46 Secondary syphilis commonly affects the palms and soles, with scaling, firm, red-brown papules. (Courtesy of Alvin Solomon, MD; with permission.)
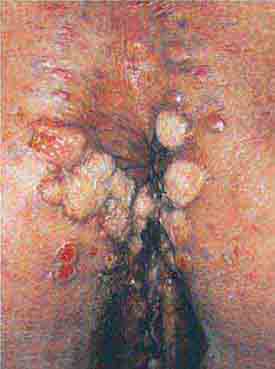
FIGURE 76e-47 Condylomata lata are moist, somewhat verrucous inter-triginous plaques seen in secondary syphilis. (Courtesy of Yale Resident’s Slide Collection; with permission.)
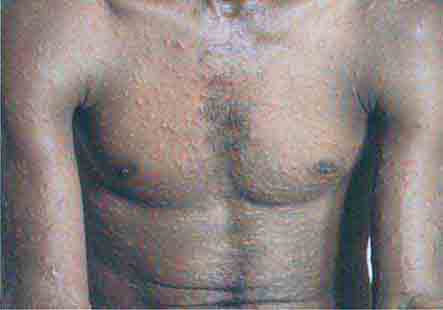
FIGURE 76e-48 Secondary syphilis, with the characteristic papulosquamous truncal eruption.
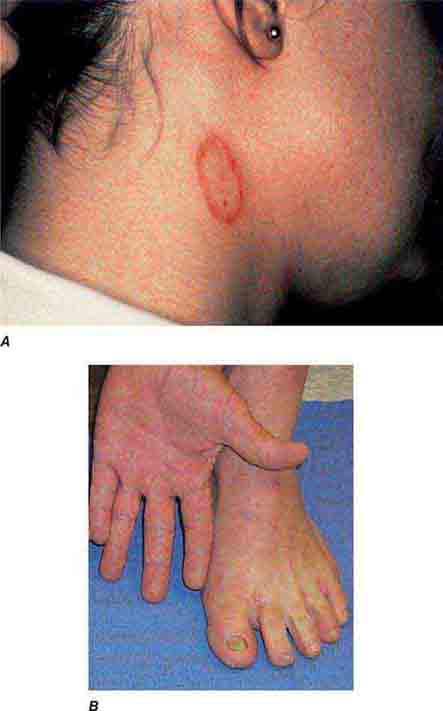
FIGURE 76e-49 A. Tinea corporis is a superficial fungal infection, seen here as an erythematous annular scaly plaque with central clearing. B. A common presentation of chronic dermatophyte infection involves the feet (tinea pedis), hands (tinea manum), and nails (tinea unguium).
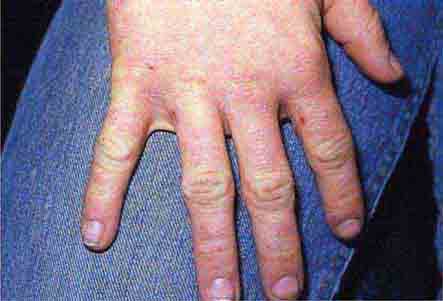
FIGURE 76e-50 Scabies, with typical scaling erythematous papules and few linear burrows.
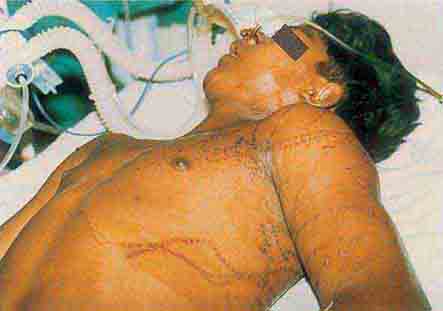
FIGURE 76e-51 Skin lesions caused by Chironex fleckeri sting. (Courtesy of V. Pranava Murthy, MD; with permission.)
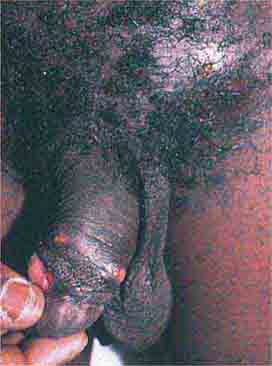
FIGURE 76e-52 Chancroid, with characteristic penile ulcers and associated left inguinal adenitis (bubo).
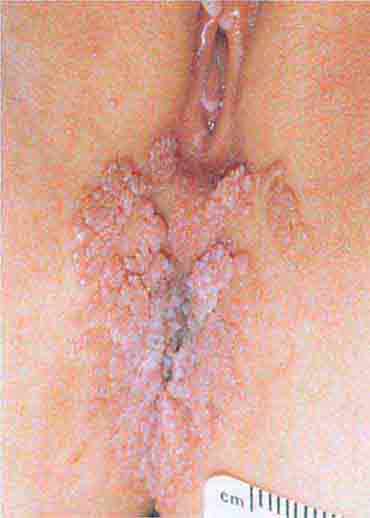
FIGURE 76e-53 Condylomata acuminata are lesions induced by human papillomavirus and in this patient are seen as multiple verrucous papules coalescing into plaques. (Courtesy of S. Wright Caughman, MD; with permission.)
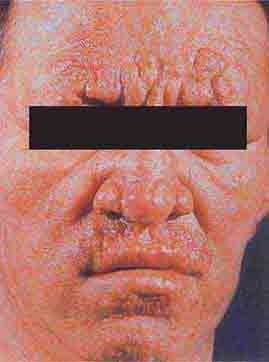
FIGURE 76e-54 A patient with features of polar lepromatous leprosy: multiple nodular skin lesions, particularly of the forehead, and loss of eyebrows. (Courtesy of Robert Gelber, MD; with permission.)
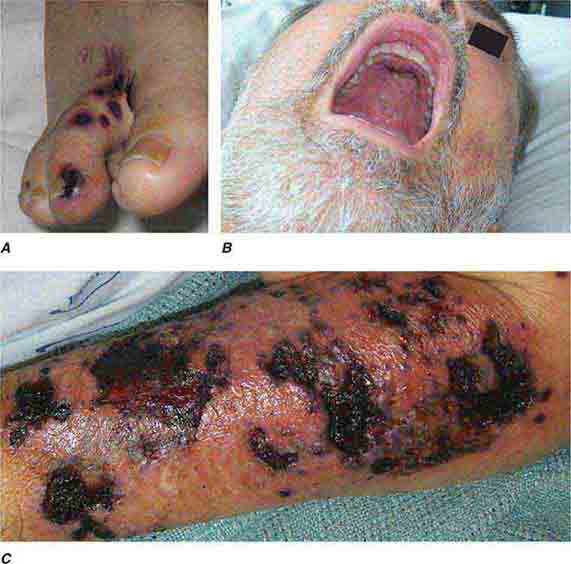
FIGURE 76e-55 Skin lesions of neutropenic patients. A. Hemorrhagic papules on the foot of a patient undergoing treatment for multiple myeloma. Biopsy and culture demonstrated Aspergillosis species. B. Eroded nodule on the hard palate of a patient undergoing chemotherapy. Biopsy and culture demonstrated Mucor species. C. Ecthyma gangrenosum in a neutropenic patient with Pseudomonas aeruginosa bacteremia.
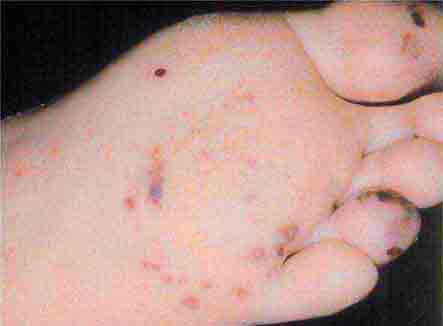
FIGURE 76e-56 Septic emboli, with hemorrhage and infarction due to acute Staphylococcus aureus endocarditis. (Courtesy of L. Baden, MD; with permission.)
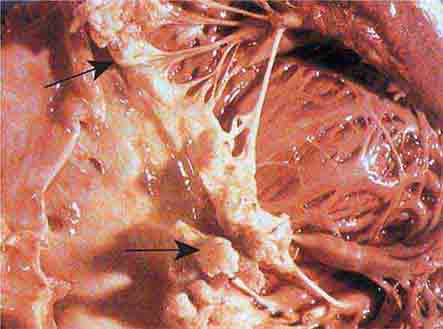
FIGURE 76e-57 Vegetations (arrows) due to viridans streptococcal endocarditis involving the mitral valve. (Courtesy of AW Karchmer, MD; with permission.)
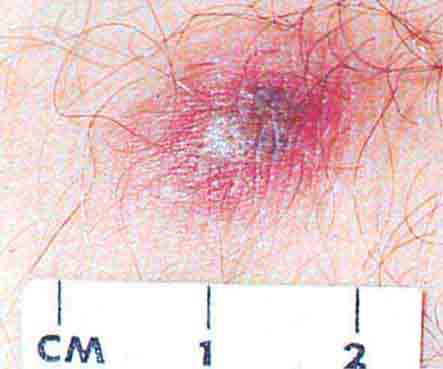
FIGURE 76e-58 Disseminated gonococcemia in the skin is seen as hemorrhagic papules and pustules with purpuric centers in an acral distribution. (Courtesy of Daniel M. Musher, MD; with permission.)
IMMUNOLOGICALLY MEDIATED SKIN DISEASE
(Figs. 76e-59 to 76e-70) Immunologically mediated skin disease may be largely localized to skin and mucous membranes and manifest with blisters and erosions such as pemphigus, pemphigoid, and dermatitis herpetiformis. In diseases such as systemic lupus erythematosus, dermatomyositis, and vasculitis, skin manifestations are often only one element of a widespread process.
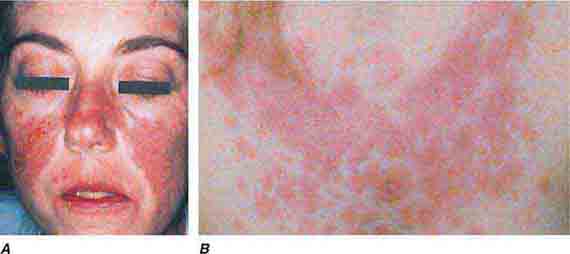
FIGURE 76e-59 Lupus erythematosus. A. Systemic lupus erythematosus, with prominent, scaly malar erythema. Involvement of other sun-exposed sites is also common. B. Acute lupus erythematosus on the upper chest, with brightly erythematous and slightly edematous coalescence of papules and plaques. (B: Courtesy of Robert Swerlick, MD; with permission.)
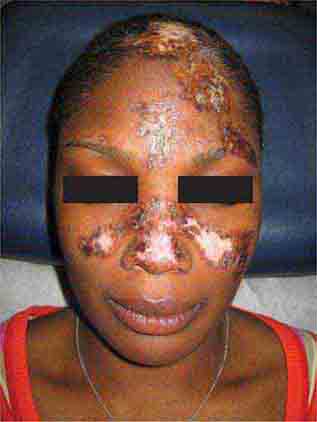
FIGURE 76e-60 Discoid lupus erythematosus. Atrophic, depigmented plaques and patches surrounded by hyperpigmentation and erythema in association with scarring and alopecia are characteristic of this cutaneous form of lupus.
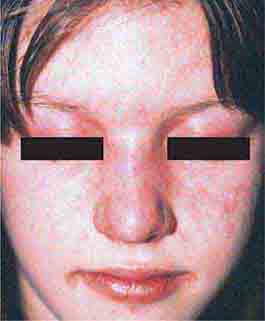
FIGURE 76e-61 Dermatomyositis. Periorbital violaceous erythema characterizes the classic heliotrope rash. (Courtesy of James Krell, MD; with permission.)
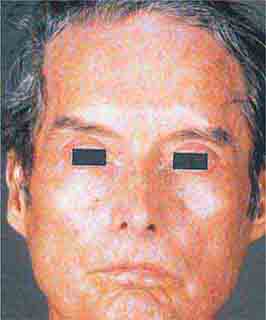
FIGURE 76e-62 Scleroderma characterized by typical expressionless, mask-like facies.
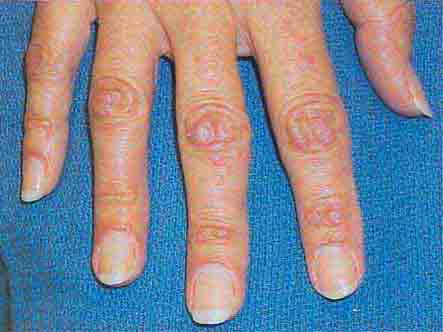
FIGURE 76e-63 Scleroderma, with acral sclerosis and focal digital ulcers.
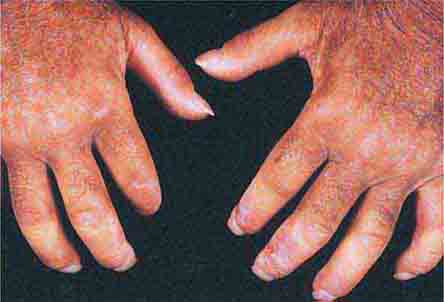
FIGURE 76e-64 Dermatomyositis often involves the hands as erythematous flat-topped papules over the knuckles (Gottron’s sign) and periungual telangiectasias.
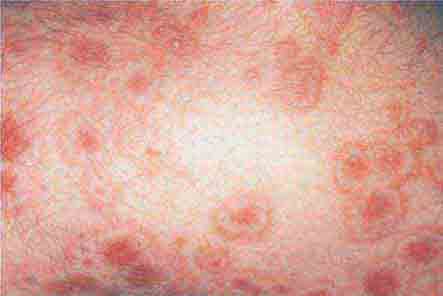
FIGURE 76e-65 Erythema multiforme is characterized by multiple erythematous plaques with a target or iris morphology and usually represents a hypersensitivity reaction to drugs or infections (especially herpes simplex virus). (Courtesy of Yale Resident’s Slide Collection; with permission.)
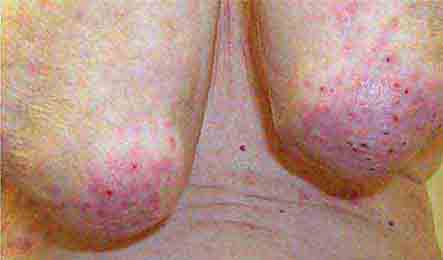
FIGURE 76e-66 Dermatitis herpetiformis, manifested by pruritic, grouped vesicles in a typical location. The vesicles are often excoriated and may also occur on the knees, buttocks, elbows, and posterior scalp.
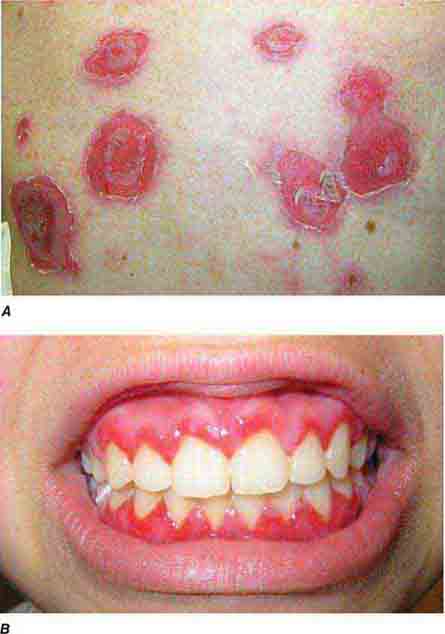
FIGURE 76e-67 Pemphigus vulgaris. A. Eroded bullae on the back. B. The oral mucosa is almost invariably involved, sometimes with erosions on the gingiva, buccal mucosa, palate, posterior pharynx, or tongue. (B: Courtesy of Robert Swerlick, MD; with permission.)
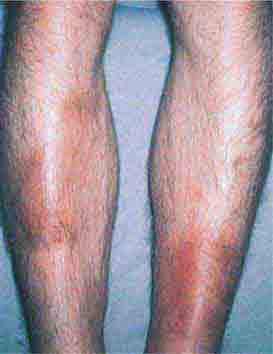
FIGURE 76e-68 Erythema nodosum is a panniculitis characterized by tender deep-seated nodules and plaques, usually located on the lower extremities. (Courtesy of Robert Swerlick, MD; with permission.)
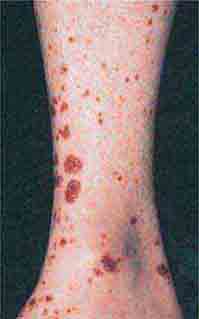
FIGURE 76e-69 Vasculitis. Palpable purpuric papules on the lower legs are seen in this patient with cutaneous small-vessel vasculitis. (Courtesy of Robert Swerlick, MD; with permission.)
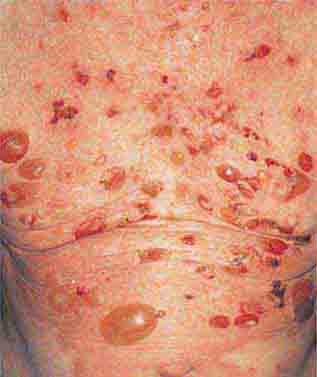
FIGURE 76e-70 Bullous pemphigoid, with tense vesicles and bullae on an erythematous, urticarial base. (Courtesy of Yale Resident’s Slide Collection; with permission.)
SKIN MANIFESTATIONS OF INTERNAL DISEASE
(Figs. 76e-71 to 76e-78) While many systemic diseases also have cutaneous manifestations, there are well-recognized dermatologic markers of internal disease, some of which are shown in this section. Many of these dermatologic markers may precede, accompany, or follow diagnosis of systemic disease. Acanthosis nigricans is a prototypical dermatologic process that often occurs in association with underlying systemic abnormalities, most commonly obesity and insulin resistance. It may also be associated with other endocrine disorders and several rare genetic syndromes. Malignant acanthosis nigricans may occur in association with several malignancies, especially adenocarcinoma of the gastrointestinal tract, lung, and breast. Other markers of internal disease in this section include pretibial myxedema, which is associated with thyroid disease, and Sweet syndrome, which may be associated with hematologic malignancies, solid tumors, infections, or inflammatory bowel disease. The skin is also involved in many systemic inflammatory diseases such as sarcoidosis, rheumatoid arthritis, and lupus erythematosus.
FIGURE 76e-71 Acanthosis nigricans, with typical hyperpigmented plaques on a velvet-like, verrucous surface on the neck.
FIGURE 76e-72 Pretibial myxedema manifesting as waxy, infiltrated plaques in a patient with Graves’ disease.
FIGURE 76e-73 Erythematous, indurated plaque of Sweet syndrome, with a pseudovesicular border. (Courtesy of Robert Swerlick, MD, with permission.)
FIGURE 76e-74 Bilateral rheumatoid nodules of the upper extremities. (Courtesy of Robert Swerlick, MD; with permission.)
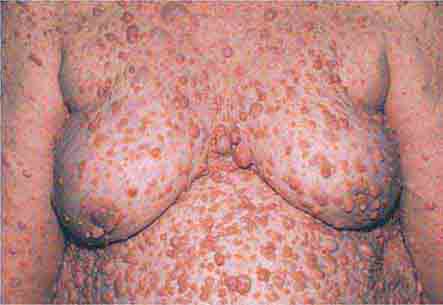
FIGURE 76e-75 Neurofibromatosis, with numerous flesh-colored cutaneous neurofibromas.
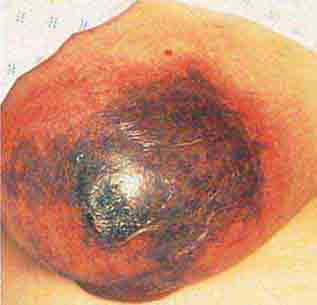
FIGURE 76e-76 Coumarin necrosis. Shown is cutaneous and subcutaneous necrosis of a breast. Other fatty areas, such as buttocks and thighs, are also common sites of involvement. (Courtesy of Kim Yancey, MD; with permission.)
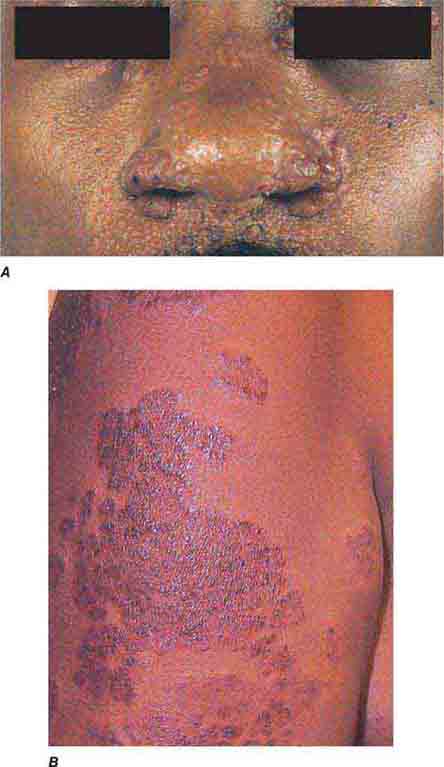
FIGURE 76e-77 Sarcoid. A. Infiltrated papules and plaques of variable color are seen in a typical paranasal and periorbital location. B. Infiltrated, hyperpigmented, and slightly erythematous coalescent papules and plaques on the upper arm. (B: Courtesy of Robert Swerlick, MD; with permission.)
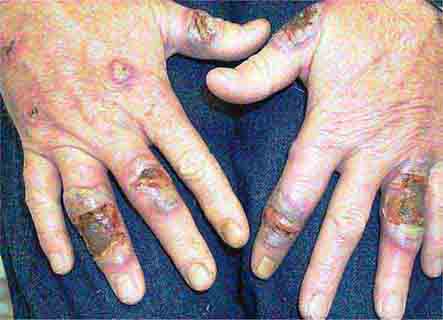
FIGURE 76e-78 Pyoderma gangrenosum on the dorsal aspect of both hands. Multiple necrotic ulcers are surrounded by a violaceous and undermined border. (Courtesy of Robert Swerlick, MD; with permission.)
SECTION 10 | HEMATOLOGIC ALTERATIONS |
77 | Anemia and Polycythemia |
HEMATOPOIESIS AND THE PHYSIOLOGIC BASIS OF RED CELL PRODUCTION
Hematopoiesis is the process by which the formed elements of blood are produced. The process is regulated through a series of steps beginning with the hematopoietic stem cell. Stem cells are capable of producing red cells, all classes of granulocytes, monocytes, platelets, and the cells of the immune system. The precise molecular mechanism—either intrinsic to the stem cell itself or through the action of extrinsic factors—by which the stem cell becomes committed to a given lineage is not fully defined. However, experiments in mice suggest that erythroid cells come from a common erythroid/megakaryocyte progenitor that does not develop in the absence of expression of the GATA-1 and FOG-1 (friend of GATA-1) transcription factors (Chap. 89e). Following lineage commitment, hematopoietic progenitor and precursor cells come increasingly under the regulatory influence of growth factors and hormones. For red cell production, erythropoietin (EPO) is the primary regulatory hormone. EPO is required for the maintenance of committed erythroid progenitor cells that, in the absence of the hormone, undergo programmed cell death (apoptosis). The regulated process of red cell production is erythropoiesis, and its key elements are illustrated in Fig. 77-1.
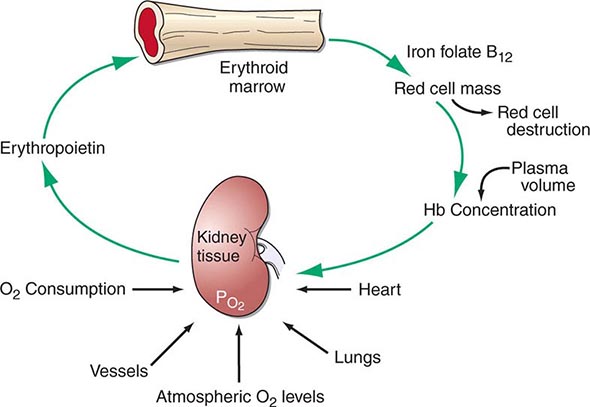
FIGURE 77-1 The physiologic regulation of red cell production by tissue oxygen tension. Hb, hemoglobin.
In the bone marrow, the first morphologically recognizable erythroid precursor is the pronormoblast. This cell can undergo four to five cell divisions, which result in the production of 16–32 mature red cells. With increased EPO production, or the administration of EPO as a drug, early progenitor cell numbers are amplified and, in turn, give rise to increased numbers of erythrocytes. The regulation of EPO production itself is linked to tissue oxygenation.
In mammals, O2 is transported to tissues bound to the hemoglobin contained within circulating red cells. The mature red cell is 8 μm in diameter, anucleate, discoid in shape, and extremely pliable in order to traverse the microcirculation successfully; its membrane integrity is maintained by the intracellular generation of ATP. Normal red cell production results in the daily replacement of 0.8–1% of all circulating red cells in the body, since the average red cell lives 100–120 days. The organ responsible for red cell production is called the erythron. The erythron is a dynamic organ made up of a rapidly proliferating pool of marrow erythroid precursor cells and a large mass of mature circulating red blood cells. The size of the red cell mass reflects the balance of red cell production and destruction. The physiologic basis of red cell production and destruction provides an understanding of the mechanisms that can lead to anemia.
The physiologic regulator of red cell production, the glycoprotein hormone EPO, is produced and released by peritubular capillary lining cells within the kidney. These cells are highly specialized epithelial-like cells. A small amount of EPO is produced by hepatocytes. The fundamental stimulus for EPO production is the availability of O2 for tissue metabolic needs. Key to EPO gene regulation is hypoxia-inducible factor (HIF)-1α. In the presence of O2, HIF-1α is hydroxylated at a key proline, allowing HIF-1α to be ubiquitinated and degraded via the proteasome pathway. If O2 becomes limiting, this critical hydroxylation step does not occur, allowing HIF-1α to partner with other proteins, translocate to the nucleus, and upregulate the expression of the EPO gene, among others.
Impaired O2 delivery to the kidney can result from a decreased red cell mass (anemia), impaired O2 loading of the hemoglobin molecule or a high O2 affinity mutant hemoglobin (hypoxemia), or, rarely, impaired blood flow to the kidney (renal artery stenosis). EPO governs the day-to-day production of red cells, and ambient levels of the hormone can be measured in the plasma by sensitive immunoassays—the normal level being 10–25 U/L. When the hemoglobin concentration falls below 100–120 g/L (10–12 g/dL), plasma EPO levels increase in proportion to the severity of the anemia (Fig. 77-2). In circulation, EPO has a half-clearance time of 6–9 h. EPO acts by binding to specific receptors on the surface of marrow erythroid precursors, inducing them to proliferate and to mature. With EPO stimulation, red cell production can increase four- to fivefold within a 1- to 2-week period, but only in the presence of adequate nutrients, especially iron. The functional capacity of the erythron, therefore, requires normal renal production of EPO, a functioning erythroid marrow, and an adequate supply of substrates for hemoglobin synthesis. A defect in any of these key components can lead to anemia. Generally, anemia is recognized in the laboratory when a patient’s hemoglobin level or hematocrit is reduced below an expected value (the normal range). The likelihood and severity of anemia are defined based on the deviation of the patient’s hemoglobin/hematocrit from values expected for age- and sex-matched normal subjects. The hemoglobin concentration in adults has a Gaussian distribution. The mean hematocrit value for adult males is 47% (standard deviation, ±7%) and that for adult females is 42% (±5%). Any single hematocrit or hemoglobin value carries with it a likelihood of associated anemia. Thus, a hematocrit of <39% in an adult male or <35% in an adult female has only about a 25% chance of being normal. Hematocrit levels are less useful than hemoglobin levels in assessing anemia because they are calculated rather than measured directly. Suspected low hemoglobin or hematocrit values are more easily interpreted if previous values for the same patient are known for comparison. The World Health Organization (WHO) defines anemia as a hemoglobin level <130 g/L (13 g/dL) in men and <120 g/L (12 g/dL) in women.
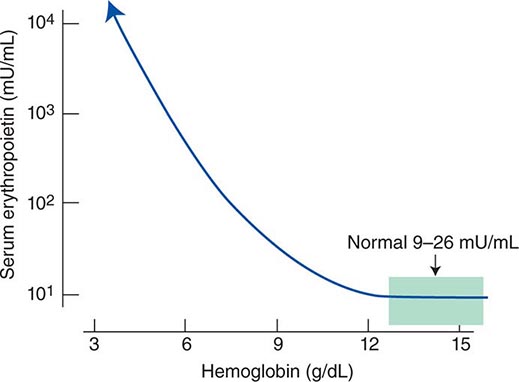
FIGURE 77-2 Erythropoietin (EPO) levels in response to anemia. When the hemoglobin level falls to 120 g/L (12 g/dL), plasma EPO levels increase logarithmically. In the presence of chronic kidney disease or chronic inflammation, EPO levels are typically lower than expected for the degree of anemia. As individuals age, the level of EPO needed to sustain normal hemoglobin levels appears to increase. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
The critical elements of erythropoiesis—EPO production, iron availability, the proliferative capacity of the bone marrow, and effective maturation of red cell precursors—are used for the initial classification of anemia (see below).
ANEMIA
CLINICAL PRESENTATION OF ANEMIA
Signs and Symptoms Anemia is most often recognized by abnormal screening laboratory tests. Patients less commonly present with advanced anemia and its attendant signs and symptoms. Acute anemia is due to blood loss or hemolysis. If blood loss is mild, enhanced O2 delivery is achieved through changes in the O2–hemoglobin dissociation curve mediated by a decreased pH or increased CO2 (Bohr effect). With acute blood loss, hypovolemia dominates the clinical picture, and the hematocrit and hemoglobin levels do not reflect the volume of blood lost. Signs of vascular instability appear with acute losses of 10–15% of the total blood volume. In such patients, the issue is not anemia but hypotension and decreased organ perfusion. When >30% of the blood volume is lost suddenly, patients are unable to compensate with the usual mechanisms of vascular contraction and changes in regional blood flow. The patient prefers to remain supine and will show postural hypotension and tachycardia. If the volume of blood lost is >40% (i.e., >2 L in the average-sized adult), signs of hypovolemic shock including confusion, dyspnea, diaphoresis, hypotension, and tachycardia appear (Chap. 129). Such patients have significant deficits in vital organ perfusion and require immediate volume replacement.
With acute hemolysis, the signs and symptoms depend on the mechanism that leads to red cell destruction. Intravascular hemolysis with release of free hemoglobin may be associated with acute back pain, free hemoglobin in the plasma and urine, and renal failure. Symptoms associated with more chronic or progressive anemia depend on the age of the patient and the adequacy of blood supply to critical organs. Symptoms associated with moderate anemia include fatigue, loss of stamina, breathlessness, and tachycardia (particularly with physical exertion). However, because of the intrinsic compensatory mechanisms that govern the O2–hemoglobin dissociation curve, the gradual onset of anemia—particularly in young patients—may not be associated with signs or symptoms until the anemia is severe (hemoglobin <70–80 g/L [7–8 g/dL]). When anemia develops over a period of days or weeks, the total blood volume is normal to slightly increased, and changes in cardiac output and regional blood flow help compensate for the overall loss in O2-carrying capacity. Changes in the position of the O2–hemoglobin dissociation curve account for some of the compensatory response to anemia. With chronic anemia, intracellular levels of 2,3-bisphosphoglycerate rise, shifting the dissociation curve to the right and facilitating O2 unloading. This compensatory mechanism can only maintain normal tissue O2 delivery in the face of a 20–30 g/L (2–3 g/dL) deficit in hemoglobin concentration. Finally, further protection of O2 delivery to vital organs is achieved by the shunting of blood away from organs that are relatively rich in blood supply, particularly the kidney, gut, and skin.
Certain disorders are commonly associated with anemia. Chronic inflammatory states (e.g., infection, rheumatoid arthritis, cancer) are associated with mild to moderate anemia, whereas lymphoproliferative disorders, such as chronic lymphocytic leukemia and certain other B cell neoplasms, may be associated with autoimmune hemolysis.
APPROACH TO THE PATIENT:
Anemia
The evaluation of the patient with anemia requires a careful history and physical examination. Nutritional history related to drugs or alcohol intake and family history of anemia should always be assessed. Certain geographic backgrounds and ethnic origins are associated with an increased likelihood of an inherited disorder of the hemoglobin molecule or intermediary metabolism. Glucose-6-phosphate dehydrogenase (G6PD) deficiency and certain hemoglobinopathies are seen more commonly in those of Middle Eastern or African origin, including African Americans who have a high frequency of G6PD deficiency. Other information that may be useful includes exposure to certain toxic agents or drugs and symptoms related to other disorders commonly associated with anemia. These include symptoms and signs such as bleeding, fatigue, malaise, fever, weight loss, night sweats, and other systemic symptoms. Clues to the mechanisms of anemia may be provided on physical examination by findings of infection, blood in the stool, lymphadenopathy, splenomegaly, or petechiae. Splenomegaly and lymphadenopathy suggest an underlying lymphoproliferative disease, whereas petechiae suggest platelet dysfunction. Past laboratory measurements are helpful to determine a time of onset.
In the anemic patient, physical examination may demonstrate a forceful heartbeat, strong peripheral pulses, and a systolic “flow” murmur. The skin and mucous membranes may be pale if the hemoglobin is <80–100 g/L (8–10 g/dL). This part of the physical examination should focus on areas where vessels are close to the surface such as the mucous membranes, nail beds, and palmar creases. If the palmar creases are lighter in color than the surrounding skin when the hand is hyperextended, the hemoglobin level is usually <80 g/L (8 g/dL).
LABORATORY EVALUATION
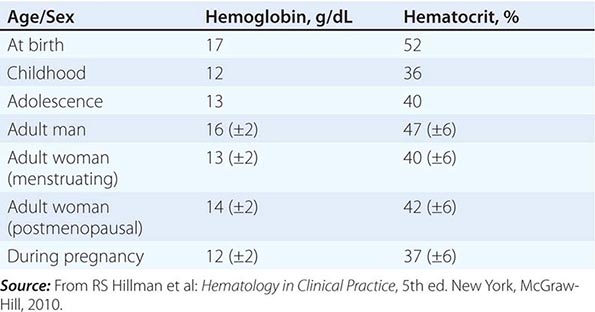
Table 77-1 lists the tests used in the initial workup of anemia. A routine complete blood count (CBC) is required as part of the evaluation and includes the hemoglobin, hematocrit, and red cell indices: the mean cell volume (MCV) in femtoliters, mean cell hemoglobin (MCH) in picograms per cell, and mean concentration of hemoglobin per volume of red cells (MCHC) in grams per liter (non-SI: grams per deciliter). The red cell indices are calculated as shown in Table 77-2, and the normal variations in the hemoglobin and hematocrit with age are shown in Table 77-3. A number of physiologic factors affect the CBC, including age, sex, pregnancy, smoking, and altitude. High-normal hemoglobin values may be seen in men and women who live at altitude or smoke heavily. Hemoglobin elevations due to smoking reflect normal compensation due to the displacement of O2 by CO in hemoglobin binding. Other important information is provided by the reticulocyte count and measurements of iron supply including serum iron, total iron-binding capacity (TIBC; an indirect measure of serum transferrin), and serum ferritin. Marked alterations in the red cell indices usually reflect disorders of maturation or iron deficiency. A careful evaluation of the peripheral blood smear is important, and clinical laboratories often provide a description of both the red and white cells, a white cell differential count, and the platelet count. In patients with severe anemia and abnormalities in red blood cell morphology and/or low reticulocyte counts, a bone marrow aspirate or biopsy can assist in the diagnosis. Other tests of value in the diagnosis of specific anemias are discussed in chapters on specific disease states.
LABORATORY TESTS IN ANEMIA DIAGNOSIS |
aM/E ratio, ratio of myeloid to erythroid precursors.
RED BLOOD CELL INDICES |

CHANGES IN NORMAL HEMOGLOBIN/HEMATOCRIT VALUES WITH AGE, SEX, AND PREGNANCY |
The components of the CBC also help in the classification of anemia. Microcytosis is reflected by a lower than normal MCV (<80), whereas high values (>100) reflect macrocytosis. The MCH and MCHC reflect defects in hemoglobin synthesis (hypochromia). Automated cell counters describe the red cell volume distribution width (RDW). The MCV (representing the peak of the distribution curve) is insensitive to the appearance of small populations of macrocytes or microcytes. An experienced laboratory technician will be able to identify minor populations of large or small cells or hypochromic cells before the red cell indices change.
Peripheral Blood Smear The peripheral blood smear provides important information about defects in red cell production (Chap. 81e). As a complement to the red cell indices, the blood smear also reveals variations in cell size (anisocytosis) and shape (poikilocytosis). The degree of anisocytosis usually correlates with increases in the RDW or the range of cell sizes. Poikilocytosis suggests a defect in the maturation of red cell precursors in the bone marrow or fragmentation of circulating red cells. The blood smear may also reveal polychromasia—red cells that are slightly larger than normal and grayish blue in color on the Wright-Giemsa stain. These cells are reticulocytes that have been prematurely released from the bone marrow, and their color represents residual amounts of ribosomal RNA. These cells appear in circulation in response to EPO stimulation or to architectural damage of the bone marrow (fibrosis, infiltration of the marrow by malignant cells, etc.) that results in their disordered release from the marrow. The appearance of nucleated red cells, Howell-Jolly bodies, target cells, sickle cells, and others may provide clues to specific disorders (Figs. 77-3 to 77-11).
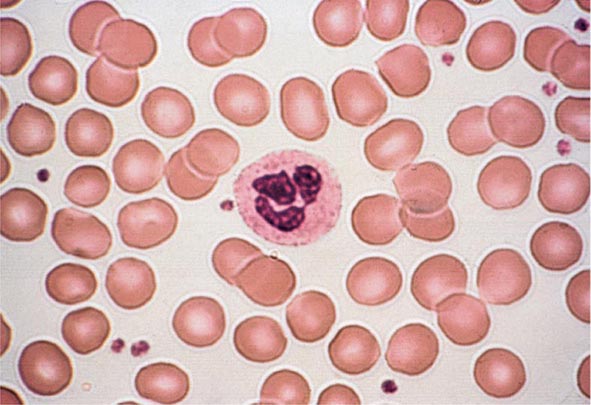
FIGURE 77-3 Normal blood smear (Wright stain). High-power field showing normal red cells, a neutrophil, and a few platelets. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
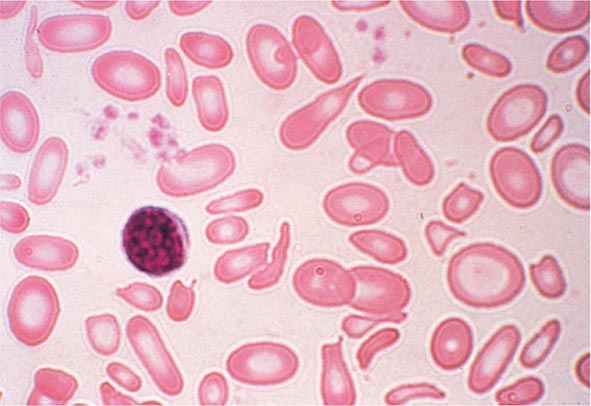
FIGURE 77-4 Severe iron-deficiency anemia. Microcytic and hypochromic red cells smaller than the nucleus of a lymphocyte associated with marked variation in size (anisocytosis) and shape (poikilocytosis). (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
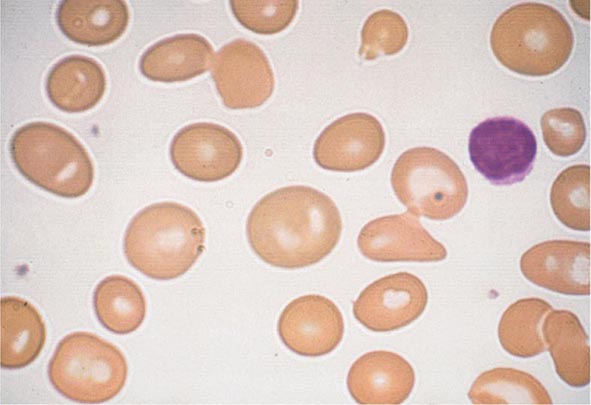
FIGURE 77-5 Macrocytosis. Red cells are larger than a small lymphocyte and well hemoglobinized. Often macrocytes are oval shaped (macro-ovalocytes).
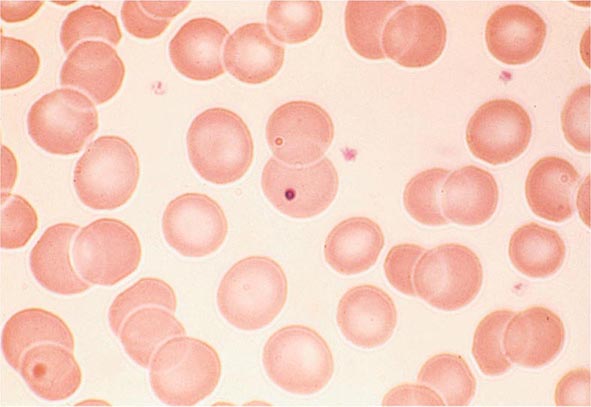
FIGURE 77-6 Howell-Jolly bodies. In the absence of a functional spleen, nuclear remnants are not culled from the red cells and remain as small homogeneously staining blue inclusions on Wright stain. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
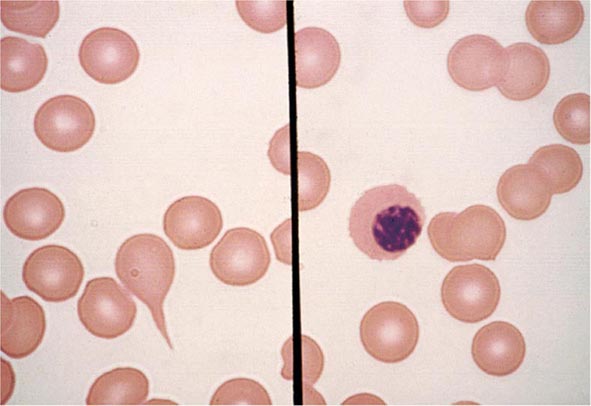
FIGURE 77-7 Red cell changes in myelofibrosis. The left panel shows a teardrop-shaped cell. The right panel shows a nucleated red cell. These forms can be seen in myelofibrosis.
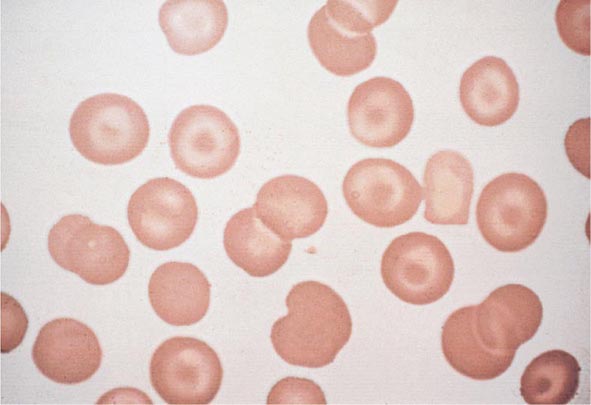
FIGURE 77-8 Target cells. Target cells have a bull’s-eye appearance and are seen in thalassemia and in liver disease. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
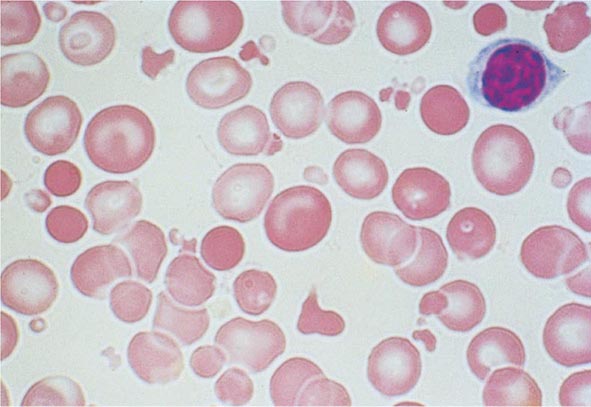
FIGURE 77-9 Red cell fragmentation. Red cells may become fragmented in the presence of foreign bodies in the circulation, such as mechanical heart valves, or in the setting of thermal injury. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
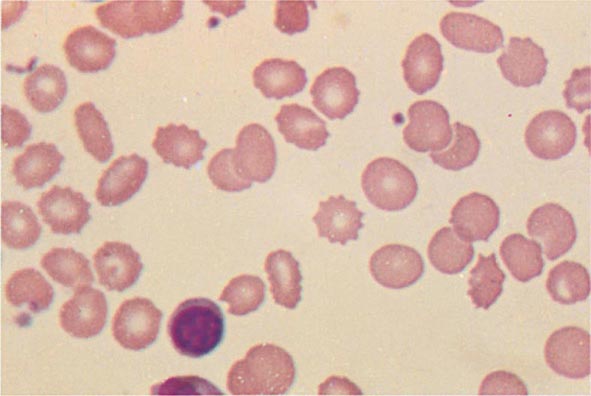
FIGURE 77-10 Uremia. The red cells in uremia may acquire numerous regularly spaced, small, spiny projections. Such cells, called burr cells or echinocytes, are readily distinguishable from irregularly spiculated acanthocytes shown in Fig. 77-11.
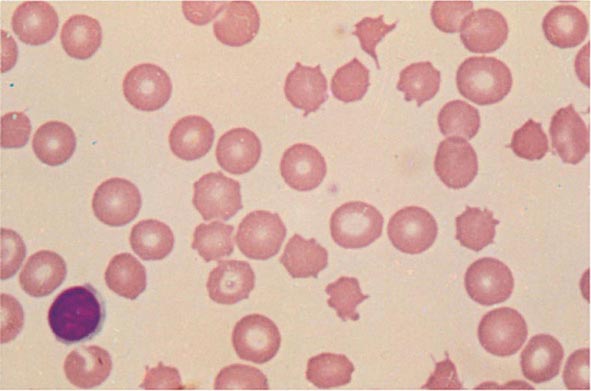
FIGURE 77-11 Spur cells. Spur cells are recognized as distorted red cells containing several irregularly distributed thornlike projections. Cells with this morphologic abnormality are also called acanthocytes. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
Reticulocyte Count An accurate reticulocyte count is key to the initial classification of anemia. Reticulocytes are red cells that have been recently released from the bone marrow. They are identified by staining with a supravital dye that precipitates the ribosomal RNA (Fig. 77-12). These precipitates appear as blue or black punctate spots and can be counted manually or, currently, by fluorescent emission of dyes that bind to RNA. This residual RNA is metabolized over the first 24–36 h of the reticulocyte’s life span in circulation. Normally, the reticulocyte count ranges from 1 to 2% and reflects the daily replacement of 0.8–1.0% of the circulating red cell population. A corrected reticulocyte count provides a reliable measure of effective red cell production.
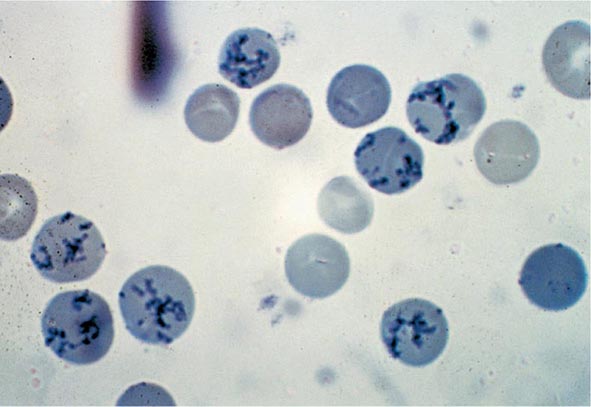
FIGURE 77-12 Reticulocytes. Methylene blue stain demonstrates residual RNA in newly made red cells. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
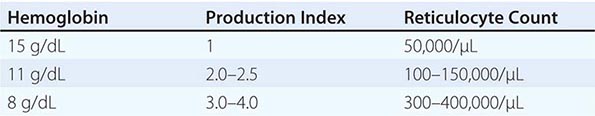
In the initial classification of anemia, the patient’s reticulocyte count is compared with the expected reticulocyte response. In general, if the EPO and erythroid marrow responses to moderate anemia [hemoglobin <100 g/L (10 g/dL)] are intact, the red cell production rate increases to two to three times normal within 10 days following the onset of anemia. In the face of established anemia, a reticulocyte response less than two to three times normal indicates an inadequate marrow response.
To use the reticulocyte count to estimate marrow response, two corrections are necessary. The first correction adjusts the reticulocyte count based on the reduced number of circulating red cells. With anemia, the percentage of reticulocytes may be increased while the absolute number is unchanged. To correct for this effect, the reticulocyte percentage is multiplied by the ratio of the patient’s hemoglobin or hematocrit to the expected hemoglobin/hematocrit for the age and sex of the patient (Table 77-4). This provides an estimate of the reticulocyte count corrected for anemia. To convert the corrected reticulocyte count to an index of marrow production, a further correction is required, depending on whether some of the reticulocytes in circulation have been released from the marrow prematurely. For this second correction, the peripheral blood smear is examined to see if there are polychromatophilic macrocytes present.
CALCULATION OF RETICULOCYTE PRODUCTION INDEX |
Correction #2 for Longer Life of Prematurely Released Reticulocytes in the Blood:
This correction produces the reticulocyte production index.
In a person whose reticulocyte count is 9%, hemoglobin 7.5 gm/dL, and hematocrit 23%, the reticulocyte production index
![]()
These cells, representing prematurely released reticulocytes, are referred to as “shift” cells, and the relationship between the degree of shift and the necessary shift correction factor is shown in Fig. 77-13. The correction is necessary because these prematurely released cells survive as reticulocytes in circulation for >1 day, thereby providing a falsely high estimate of daily red cell production. If polychromasia is increased, the reticulocyte count, already corrected for anemia, should be divided again by 2 to account for the prolonged reticulocyte maturation time. The second correction factor varies from 1 to 3 depending on the severity of anemia. In general, a correction of 2 is simply used. An appropriate correction is shown in Table 77-4. If polychromatophilic cells are not seen on the blood smear, the second correction is not required. The now doubly corrected reticulocyte count is the reticulocyte production index, and it provides an estimate of marrow production relative to normal. In many hospital laboratories, the reticulocyte count is reported not only as a percentage but also in absolute numbers. If so, no correction for dilution is required. A summary of the appropriate marrow response to varying degrees of anemia is shown in Table 77-5.
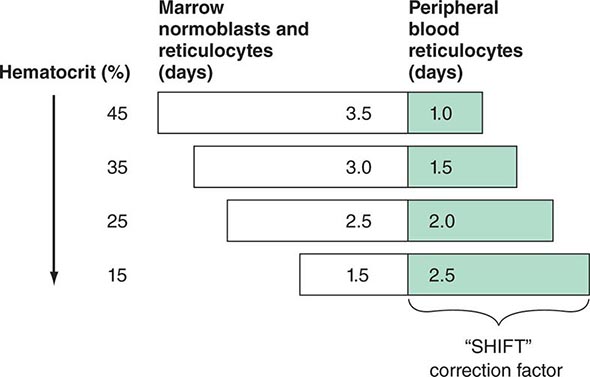
FIGURE 77-13 Correction of the reticulocyte count. To use the reticulocyte count as an indicator of effective red cell production, the reticulocyte percentage must be corrected based on the level of anemia and the circulating life span of the reticulocytes. Erythroid cells take ~4.5 days to mature. At a normal hemoglobin, reticulocytes are released to the circulation with ~1 day left as reticulocytes. However, with different levels of anemia, reticulocytes (and even earlier erythroid cells) may be released from the marrow prematurely. Most patients come to clinical attention with hematocrits in the mid-20s, and thus a correction factor of 2 is commonly used because the observed reticulocytes will live for 2 days in the circulation before losing their RNA.
NORMAL MARROW RESPONSE TO ANEMIA |
Premature release of reticulocytes is normally due to increased EPO stimulation. However, if the integrity of the bone marrow release process is lost through tumor infiltration, fibrosis, or other disorders, the appearance of nucleated red cells or polychromatophilic macrocytes should still invoke the second reticulocyte correction. The shift correction should always be applied to a patient with anemia and a very high reticulocyte count to provide a true index of effective red cell production. Patients with severe chronic hemolytic anemia may increase red cell production as much as six- to sevenfold. This measure alone confirms the fact that the patient has an appropriate EPO response, a normally functioning bone marrow, and sufficient iron available to meet the demands for new red cell formation. If the reticulocyte production index is <2 in the face of established anemia, a defect in erythroid marrow proliferation or maturation must be present.
Tests of Iron Supply and Storage The laboratory measurements that reflect the availability of iron for hemoglobin synthesis include the serum iron, the TIBC, and the percent transferrin saturation. The percent transferrin saturation is derived by dividing the serum iron level (× 100) by the TIBC. The normal serum iron ranges from 9 to 27 μmol/L (50–150 μg/dL), whereas the normal TIBC is 54–64 μmol/L (300–360 μg/dL); the normal transferrin saturation ranges from 25 to 50%. A diurnal variation in the serum iron leads to a variation in the percent transferrin saturation. The serum ferritin is used to evaluate total body iron stores. Adult males have serum ferritin levels that average ~100 μg/L, corresponding to iron stores of ~1 g. Adult females have lower serum ferritin levels averaging 30 μg/L, reflecting lower iron stores (~300 mg). A serum ferritin level of 10–15 μg/L indicates depletion of body iron stores. However, ferritin is also an acute-phase reactant and, in the presence of acute or chronic inflammation, may rise several-fold above baseline levels. As a rule, a serum ferritin >200 μg/L means there is at least some iron in tissue stores.
Bone Marrow Examination A bone marrow aspirate and smear or a needle biopsy can be useful in the evaluation of some patients with anemia. In patients with hypoproliferative anemia and normal iron status, a bone marrow is indicated. Marrow examination can diagnose primary marrow disorders such as myelofibrosis, a red cell maturation defect, or an infiltrative disease (Figs. 77-14 to 77-16). The increase or decrease of one cell lineage (myeloid vs erythroid) compared to another is obtained by a differential count of nucleated cells in a bone marrow smear (the myeloid/erythroid [M/E] ratio). A patient with a hypoproliferative anemia (see below) and a reticulocyte production index <2 will demonstrate an M/E ratio of 2 or 3:1. In contrast, patients with hemolytic disease and a production index >3 will have an M/E ratio of at least 1:1. Maturation disorders are identified from the discrepancy between the M/E ratio and the reticulocyte production index (see below). Either the marrow smear or biopsy can be stained for the presence of iron stores or iron in developing red cells. The storage iron is in the form of ferritin or hemosiderin. On carefully prepared bone marrow smears, small ferritin granules can normally be seen under oil immersion in 20–40% of developing erythroblasts. Such cells are called sideroblasts.
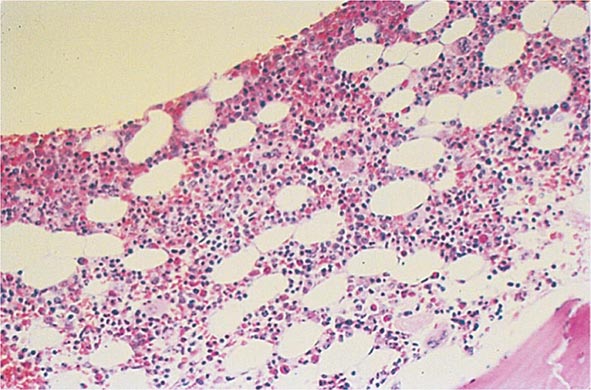
FIGURE 77-14 Normal bone marrow. This is a low-power view of a section of a normal bone marrow biopsy stained with hematoxylin and eosin (H&E). Note that the nucleated cellular elements account for ~40–50% and the fat (clear areas) accounts for ~50–60% of the area. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
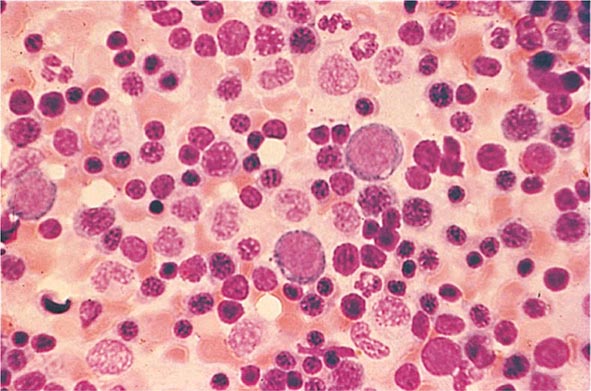
FIGURE 77-15 Erythroid hyperplasia. This marrow shows an increase in the fraction of cells in the erythroid lineage as might be seen when a normal marrow compensates for acute blood loss or hemolysis. The myeloid/erythroid (M/E) ratio is about 1:1. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
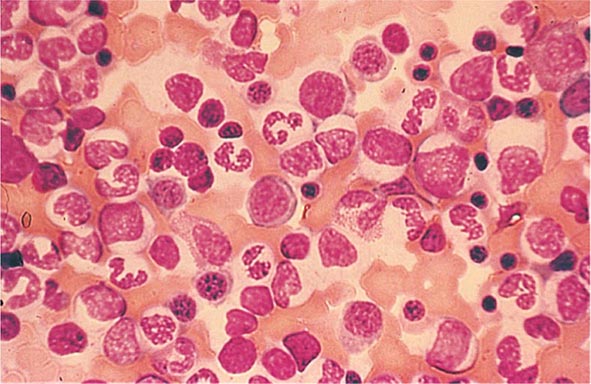
FIGURE 77-16 Myeloid hyperplasia. This marrow shows an increase in the fraction of cells in the myeloid or granulocytic lineage as might be seen in a normal marrow responding to infection. The myeloid/erythroid (M/E) ratio is >3:1. (From RS Hillman et al: Hematology in Clinical Practice, 5th ed. New York, McGraw-Hill, 2010.)
Additional laboratory tests may be of value in confirming specific diagnoses. For details of these tests and how they are applied in individual disorders, see Chaps. 126 to 130.
DEFINITION AND CLASSIFICATION OF ANEMIA
Initial Classification of Anemia The functional classification of anemia has three major categories. These are (1) marrow production defects (hypoproliferation), (2) red cell maturation defects (ineffective erythropoiesis), and (3) decreased red cell survival (blood loss/hemolysis). The classification is shown in Fig. 77-17. A hypoproliferative anemia is typically seen with a low reticulocyte production index together with little or no change in red cell morphology (a normocytic, normochromic anemia) (Chap. 126). Maturation disorders typically have a slight to moderately elevated reticulocyte production index that is accompanied by either macrocytic (Chap. 128) or microcytic (Chaps. 126, 127) red cell indices. Increased red blood cell destruction secondary to hemolysis results in an increase in the reticulocyte production index to at least three times normal (Chap. 129), provided sufficient iron is available. Hemorrhagic anemia does not typically result in production indices of more than 2.0–2.5 times normal because of the limitations placed on expansion of the erythroid marrow by iron availability.
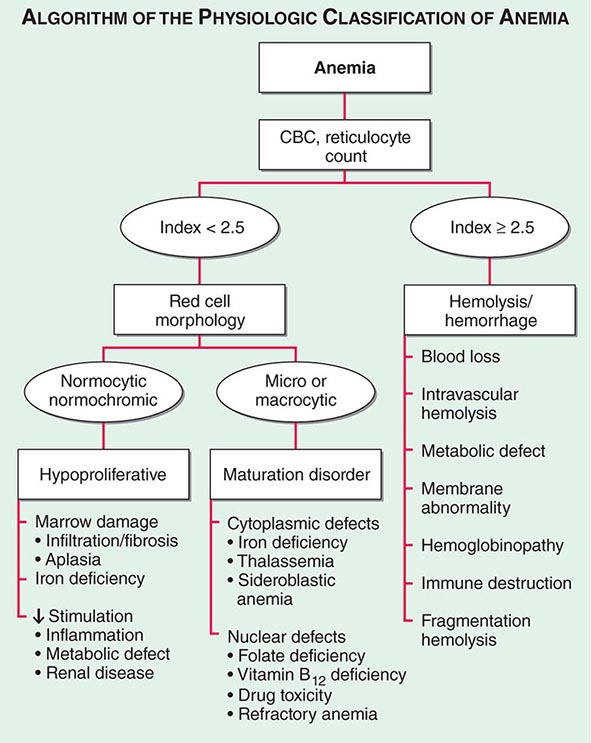
FIGURE 77-17 The physiologic classification of anemia. CBC, complete blood count.
In the first branch point of the classification of anemia, a reticulocyte production index >2.5 indicates that hemolysis is most likely. A reticulocyte production index <2 indicates either a hypoproliferative anemia or maturation disorder. The latter two possibilities can often be distinguished by the red cell indices, by examination of the peripheral blood smear, or by a marrow examination. If the red cell indices are normal, the anemia is almost certainly hypoproliferative in nature. Maturation disorders are characterized by ineffective red cell production and a low reticulocyte production index. Bizarre red cell shapes—macrocytes or hypochromic microcytes—are seen on the peripheral blood smear. With a hypoproliferative anemia, no erythroid hyperplasia is noted in the marrow, whereas patients with ineffective red cell production have erythroid hyperplasia and an M/E ratio <1:1.
Hypoproliferative Anemias At least 75% of all cases of anemia are hypoproliferative in nature. A hypoproliferative anemia reflects absolute or relative marrow failure in which the erythroid marrow has not proliferated appropriately for the degree of anemia. The majority of hypoproliferative anemias are due to mild to moderate iron deficiency or inflammation. A hypoproliferative anemia can result from marrow damage, iron deficiency, or inadequate EPO stimulation. The last may reflect impaired renal function, suppression of EPO production by inflammatory cytokines such as interleukin 1, or reduced tissue needs for O2 from metabolic disease such as hypothyroidism. Only occasionally is the marrow unable to produce red cells at a normal rate, and this is most prevalent in patients with renal failure. With diabetes mellitus or myeloma, the EPO deficiency may be more marked than would be predicted by the degree of renal insufficiency. In general, hypoproliferative anemias are characterized by normocytic, normochromic red cells, although microcytic, hypochromic cells may be observed with mild iron deficiency or long-standing chronic inflammatory disease. The key laboratory tests in distinguishing between the various forms of hypoproliferative anemia include the serum iron and iron-binding capacity, evaluation of renal and thyroid function, a marrow biopsy or aspirate to detect marrow damage or infiltrative disease, and serum ferritin to assess iron stores. An iron stain of the marrow will determine the pattern of iron distribution. Patients with the anemia of acute or chronic inflammation show a distinctive pattern of serum iron (low), TIBC (normal or low), percent transferrin saturation (low), and serum ferritin (normal or high). These changes in iron values are brought about by hepcidin, the iron regulatory hormone that is produced by the liver and is increased in inflammation (Chap. 126). A distinct pattern of results is noted in mild to moderate iron deficiency (low serum iron, high TIBC, low percent transferrin saturation, low serum ferritin) (Chap. 126). Marrow damage by drugs, infiltrative disease such as leukemia or lymphoma, or marrow aplasia is diagnosed from the peripheral blood and bone marrow morphology. With infiltrative disease or fibrosis, a marrow biopsy is required.
Maturation Disorders The presence of anemia with an inappropriately low reticulocyte production index, macro- or microcytosis on smear, and abnormal red cell indices suggests a maturation disorder. Maturation disorders are divided into two categories: nuclear maturation defects, associated with macrocytosis, and cytoplasmic maturation defects, associated with microcytosis and hypochromia usually from defects in hemoglobin synthesis. The inappropriately low reticulocyte production index is a reflection of the ineffective erythropoiesis that results from the destruction within the marrow of developing erythroblasts. Bone marrow examination shows erythroid hyperplasia.
Nuclear maturation defects result from vitamin B12 or folic acid deficiency, drug damage, or myelodysplasia. Drugs that interfere with cellular DNA synthesis, such as methotrexate or alkylating agents, can produce a nuclear maturation defect. Alcohol, alone, is also capable of producing macrocytosis and a variable degree of anemia, but this is usually associated with folic acid deficiency. Measurements of folic acid and vitamin B12 are critical not only in identifying the specific vitamin deficiency but also because they reflect different pathogenetic mechanisms (Chap. 128).
Cytoplasmic maturation defects result from severe iron deficiency or abnormalities in globin or heme synthesis. Iron deficiency occupies an unusual position in the classification of anemia. If the iron-deficiency anemia is mild to moderate, erythroid marrow proliferation is blunted and the anemia is classified as hypoproliferative. However, if the anemia is severe and prolonged, the erythroid marrow will become hyperplastic despite the inadequate iron supply, and the anemia will be classified as ineffective erythropoiesis with a cytoplasmic maturation defect. In either case, an inappropriately low reticulocyte production index, microcytosis, and a classic pattern of iron values make the diagnosis clear and easily distinguish iron deficiency from other cytoplasmic maturation defects such as the thalassemias. Defects in heme synthesis, in contrast to globin synthesis, are less common and may be acquired or inherited (Chap. 430). Acquired abnormalities are usually associated with myelodysplasia, may lead to either a macro- or microcytic anemia, and are frequently associated with mitochondrial iron loading. In these cases, iron is taken up by the mitochondria of the developing erythroid cell but not incorporated into heme. The iron-encrusted mitochondria surround the nucleus of the erythroid cell, forming a ring. Based on the distinctive finding of so-called ringed sideroblasts on the marrow iron stain, patients are diagnosed as having a sideroblastic anemia—almost always reflecting myelodysplasia. Again, studies of iron parameters are helpful in the differential diagnosis of these patients.
Blood Loss/Hemolytic Anemia In contrast to anemias associated with an inappropriately low reticulocyte production index, hemolysis is associated with red cell production indices ≥2.5 times normal. The stimulated erythropoiesis is reflected in the blood smear by the appearance of increased numbers of polychromatophilic macrocytes. A marrow examination is rarely indicated if the reticulocyte production index is increased appropriately. The red cell indices are typically normocytic or slightly macrocytic, reflecting the increased number of reticulocytes. Acute blood loss is not associated with an increased reticulocyte production index because of the time required to increase EPO production and, subsequently, marrow proliferation. Subacute blood loss may be associated with modest reticulocytosis. Anemia from chronic blood loss presents more often as iron deficiency than with the picture of increased red cell production.
The evaluation of blood loss anemia is usually not difficult. Most problems arise when a patient presents with an increased red cell production index from an episode of acute blood loss that went unrecognized. The cause of the anemia and increased red cell production may not be obvious. The confirmation of a recovering state may require observations over a period of 2–3 weeks, during which the hemoglobin concentration will rise and the reticulocyte production index fall (Chap. 129).
Hemolytic disease, while dramatic, is among the least common forms of anemia. The ability to sustain a high reticulocyte production index reflects the ability of the erythroid marrow to compensate for hemolysis and, in the case of extravascular hemolysis, the efficient recycling of iron from the destroyed red cells to support red cell production. With intravascular hemolysis, such as paroxysmal nocturnal hemoglobinuria, the loss of iron may limit the marrow response. The level of response depends on the severity of the anemia and the nature of the underlying disease process.
Hemoglobinopathies, such as sickle cell disease and the thalassemias, present a mixed picture. The reticulocyte index may be high but is inappropriately low for the degree of marrow erythroid hyperplasia (Chap. 127).
Hemolytic anemias present in different ways. Some appear suddenly as an acute, self-limited episode of intravascular or extravascular hemolysis, a presentation pattern often seen in patients with autoimmune hemolysis or with inherited defects of the Embden-Meyerhof pathway or the glutathione reductase pathway. Patients with inherited disorders of the hemoglobin molecule or red cell membrane generally have a lifelong clinical history typical of the disease process. Those with chronic hemolytic disease, such as hereditary spherocytosis, may actually present not with anemia but with a complication stemming from the prolonged increase in red cell destruction such as symptomatic bilirubin gallstones or splenomegaly. Patients with chronic hemolysis are also susceptible to aplastic crises if an infectious process interrupts red cell production.
The differential diagnosis of an acute or chronic hemolytic event requires the careful integration of family history, the pattern of clinical presentation, and—whether the disease is congenital or acquired—careful examination of the peripheral blood smear. Precise diagnosis may require more specialized laboratory tests, such as hemoglobin electrophoresis or a screen for red cell enzymes. Acquired defects in red cell survival are often immunologically mediated and require a direct or indirect antiglobulin test or a cold agglutinin titer to detect the presence of hemolytic antibodies or complement-mediated red cell destruction (Chap. 129).
POLYCYTHEMIA
Polycythemia is defined as an increase in the hemoglobin above normal. This increase may be real or only apparent because of a decrease in plasma volume (spurious or relative polycythemia). The term erythrocytosis may be used interchangeably with polycythemia, but some draw a distinction between them: erythrocytosis implies documentation of increased red cell mass, whereas polycythemia refers to any increase in red cells. Often patients with polycythemia are detected through an incidental finding of elevated hemoglobin or hematocrit levels. Concern that the hemoglobin level may be abnormally high is usually triggered at 170 g/L (17 g/dL) for men and 150 g/L (15 g/dL) for women. Hematocrit levels >50% in men or >45% in women may be abnormal. Hematocrits >60% in men and >55% in women are almost invariably associated with an increased red cell mass. Given that the machine that quantitates red cell parameters actually measures hemoglobin concentrations and calculates hematocrits, hemoglobin levels may be a better index.
Features of the clinical history that are useful in the differential diagnosis include smoking history; current living at high altitude; or a history of congenital heart disease, sleep apnea, or chronic lung disease.
Patients with polycythemia may be asymptomatic or experience symptoms related to the increased red cell mass or the underlying disease process that leads to the increased red cell mass. The dominant symptoms from an increased red cell mass are related to hyperviscosity and thrombosis (both venous and arterial), because the blood viscosity increases logarithmically at hematocrits >55%. Manifestations range from digital ischemia to Budd-Chiari syndrome with hepatic vein thrombosis. Abdominal vessel thromboses are particularly common. Neurologic symptoms such as vertigo, tinnitus, headache, and visual disturbances may occur. Hypertension is often present. Patients with polycythemia vera may have aquagenic pruritus and symptoms related to hepatosplenomegaly. Patients may have easy bruising, epistaxis, or bleeding from the gastrointestinal tract. Peptic ulcer disease is common. Patients with hypoxemia may develop cyanosis on minimal exertion or have headache, impaired mental acuity, and fatigue.
The physical examination usually reveals a ruddy complexion. Splenomegaly favors polycythemia vera as the diagnosis (Chap. 131). The presence of cyanosis or evidence of a right-to-left shunt suggests congenital heart disease presenting in the adult, particularly tetralogy of Fallot or Eisenmenger’s syndrome (Chap. 236). Increased blood viscosity raises pulmonary artery pressure; hypoxemia can lead to increased pulmonary vascular resistance. Together, these factors can produce cor pulmonale.
Polycythemia can be spurious (related to a decrease in plasma volume; Gaisbock’s syndrome), primary, or secondary in origin. The secondary causes are all associated with increases in EPO levels: either a physiologically adapted appropriate elevation based on tissue hypoxia (lung disease, high altitude, CO poisoning, high-affinity hemoglobinopathy) or an abnormal overproduction (renal cysts, renal artery stenosis, tumors with ectopic EPO production). A rare familial form of polycythemia is associated with normal EPO levels but hyperresponsive EPO receptors due to mutations.
APPROACH TO THE PATIENT:
Polycythemia
As shown in Fig. 77-18, the first step is to document the presence of an increased red cell mass using the principle of isotope dilution by administering 51Cr-labeled autologous red blood cells to the patient and sampling blood radioactivity over a 2-h period. If the red cell mass is normal (<36 mL/kg in men, <32 mL/kg in women), the patient has spurious or relative polycythemia. If the red cell mass is increased (>36 mL/kg in men, >32 mL/kg in women), serum EPO levels should be measured. If EPO levels are low or unmeasurable, the patient most likely has polycythemia vera. A mutation in JAK2 (Val617Phe), a key member of the cytokine intracellular signaling pathway, can be found in 90–95% of patients with polycythemia vera. Many of those without this particular JAK2 mutation have mutations in exon 12. As a practical matter, few centers assess red cell mass in the setting of an increased hematocrit. The short workup is to measure EPO levels, check for JAK2 mutation, and perform an abdominal ultrasound to assess spleen size. Tests that support the diagnosis of polycythemia vera include elevated white blood cell count, increased absolute basophil count, and thrombocytosis.
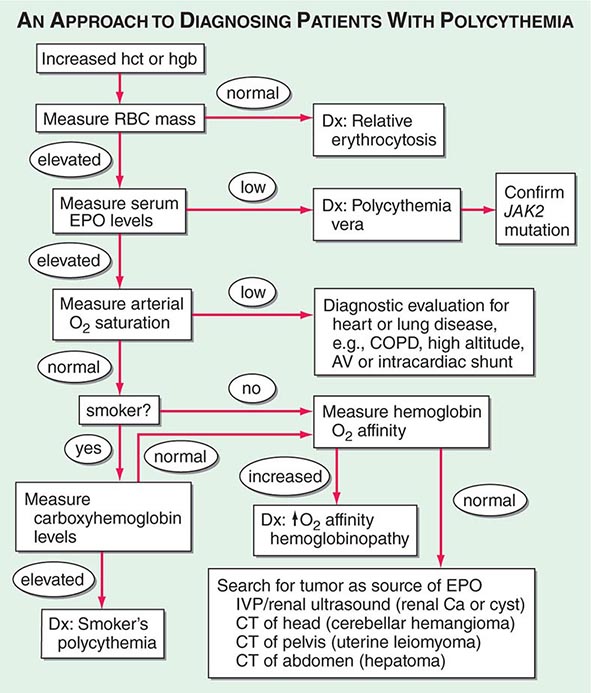
FIGURE 77-18 An approach to the differential diagnosis of patients with an elevated hemoglobin (possible polycythemia). AV, atrioventricular; COPD, chronic obstructive pulmonary disease; CT, computed tomography; EPO, erythropoietin; hct, hematocrit; hgb, hemoglobin; IVP, intravenous pyelogram; RBC, red blood cell.
If serum EPO levels are elevated, one needs to distinguish whether the elevation is a physiologic response to hypoxia or related to autonomous EPO production. Patients with low arterial O2 saturation (<92%) should be further evaluated for the presence of heart or lung disease, if they are not living at high altitude. Patients with normal O2 saturation who are smokers may have elevated EPO levels because of CO displacement of O2. If carboxyhemoglobin (COHb) levels are high, the diagnosis is “smoker’s polycythemia.” Such patients should be urged to stop smoking. Those who cannot stop smoking require phlebotomy to control their polycythemia. Patients with normal O2 saturation who do not smoke either have an abnormal hemoglobin that does not deliver O2 to the tissues (evaluated by finding elevated O2–hemoglobin affinity) or have a source of EPO production that is not responding to the normal feedback inhibition. Further workup is dictated by the differential diagnosis of EPO-producing neoplasms. Hepatoma, uterine leiomyoma, and renal cancer or cysts are all detectable with abdominopelvic computed tomography scans. Cerebellar hemangiomas may produce EPO, but they present with localizing neurologic signs and symptoms rather than polycythemia-related symptoms.
78 | Bleeding and Thrombosis |
The human hemostatic system provides a natural balance between procoagulant and anticoagulant forces. The procoagulant forces include platelet adhesion and aggregation and fibrin clot formation; anticoagulant forces include the natural inhibitors of coagulation and fibrinolysis. Under normal circumstances, hemostasis is regulated to promote blood flow; however, it is also prepared to clot blood rapidly to arrest blood flow and prevent exsanguination. After bleeding is successfully halted, the system remodels the damaged vessel to restore normal blood flow. The major components of the hemostatic system, which function in concert, are (1) platelets and other formed elements of blood, such as monocytes and red cells; (2) plasma proteins (the coagulation and fibrinolytic factors and inhibitors); and (3) the vessel wall.
STEPS OF NORMAL HEMOSTASIS
PLATELET PLUG FORMATION
On vascular injury, platelets adhere to the site of injury, usually the denuded vascular intimal surface. Platelet adhesion is mediated primarily by Von Willebrand factor (VWF), a large multimeric protein present in both plasma and the extracellular matrix of the subendothelial vessel wall, which serves as the primary “molecular glue,” providing sufficient strength to withstand the high levels of shear stress that would tend to detach them with the flow of blood. Platelet adhesion is also facilitated by direct binding to subendothelial collagen through specific platelet membrane collagen receptors.
Platelet adhesion results in subsequent platelet activation and aggregation. This process is enhanced and amplified by humoral mediators in plasma (e.g., epinephrine, thrombin); mediators released from activated platelets (e.g., adenosine diphosphate, serotonin); and vessel wall extracellular matrix constituents that come in contact with adherent platelets (e.g., collagen, VWF). Activated platelets undergo the release reaction, during which they secrete contents that further promote aggregation and inhibit the naturally anticoagulant endothelial cell factors. During platelet aggregation (platelet-platelet interaction), additional platelets are recruited from the circulation to the site of vascular injury, leading to the formation of an occlusive platelet thrombus. The platelet plug is anchored and stabilized by the developing fibrin mesh.
The platelet glycoprotein (Gp) IIb/IIIa (αIIbβ3) complex is the most abundant receptor on the platelet surface. Platelet activation converts the normally inactive Gp IIb/IIIa receptor into an active receptor, enabling binding to fibrinogen and VWF. Because the surface of each platelet has about 50,000 Gp IIb/IIIa–binding sites, numerous activated platelets recruited to the site of vascular injury can rapidly form an occlusive aggregate by means of a dense network of intercellular fibrinogen bridges. Because this receptor is the key mediator of platelet aggregation, it has become an effective target for antiplatelet therapy.
FIBRIN CLOT FORMATION
Plasma coagulation proteins (clotting factors) normally circulate in plasma in their inactive forms. The sequence of coagulation protein reactions that culminate in the formation of fibrin was originally described as a waterfall or a cascade. Two pathways of blood coagulation have been described in the past: the so-called extrinsic, or tissue factor, pathway and the so-called intrinsic, or contact activation, pathway. We now know that coagulation is normally initiated through tissue factor (TF) exposure and activation through the classic extrinsic pathway but with critically important amplification through elements of the classic intrinsic pathway, as illustrated in Fig. 78-1. These reactions take place on phospholipid surfaces, usually the activated platelet surface. Coagulation testing in the laboratory can reflect other influences due to the artificial nature of the in vitro systems used (see below).
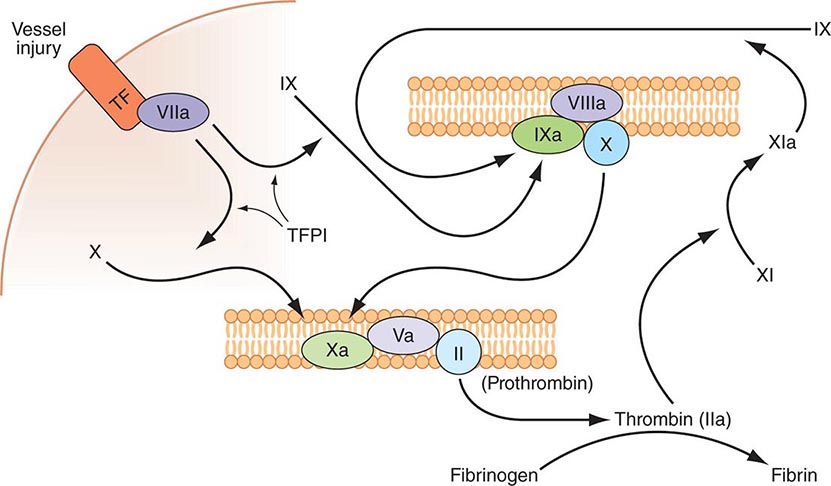
FIGURE 78-1 Coagulation is initiated by tissue factor (TF) exposure, which, with factor (F) VIIa, activates FIX and FX, which in turn, with FVIII and FV as cofactors, respectively, results in thrombin formation and subsequent conversion of fibrinogen to fibrin. Thrombin activates FXI, FVIII, and FV, amplifying the coagulation signal. Once the TF/FVIIa/FXa complex is formed, tissue factor pathway inhibitor (TFPI) inhibits the TF/FVIIa pathway, making coagulation dependent on the amplification loop through FIX/FVIII. Coagulation requires calcium (not shown) and takes place on phospholipid surfaces, usually the activated platelet membrane.
The immediate trigger for coagulation is vascular damage that exposes blood to TF that is constitutively expressed on the surfaces of subendothelial cellular components of the vessel wall, such as smooth muscle cells and fibroblasts. TF is also present in circulating microparticles, presumably shed from cells including monocytes and platelets. TF binds the serine protease factor VIIa; the complex activates factor × to factor Xa. Alternatively, the complex can indirectly activate factor × by initially converting factor IX to factor IXa, which then activates factor X. The participation of factor XI in hemostasis is not dependent on its activation by factor XIIa but rather on its positive feedback activation by thrombin. Thus, factor XIa functions in the propagation and amplification, rather than in the initiation, of the coagulation cascade.
Factor Xa can be formed through the actions of either the TF/factor VIIa complex or factor IXa (with factor VIIIa as a cofactor) and converts prothrombin to thrombin, the pivotal protease of the coagulation system. The essential cofactor for this reaction is factor Va. Like the homologous factor VIIIa, factor Va is produced by thrombin-induced limited proteolysis of factor V. Thrombin is a multifunctional enzyme that converts soluble plasma fibrinogen to an insoluble fibrin matrix. Fibrin polymerization involves an orderly process of intermolecular associations (Fig. 78-2). Thrombin also activates factor XIII (fibrin-stabilizing factor) to factor XIIIa, which covalently cross-links and thereby stabilizes the fibrin clot.
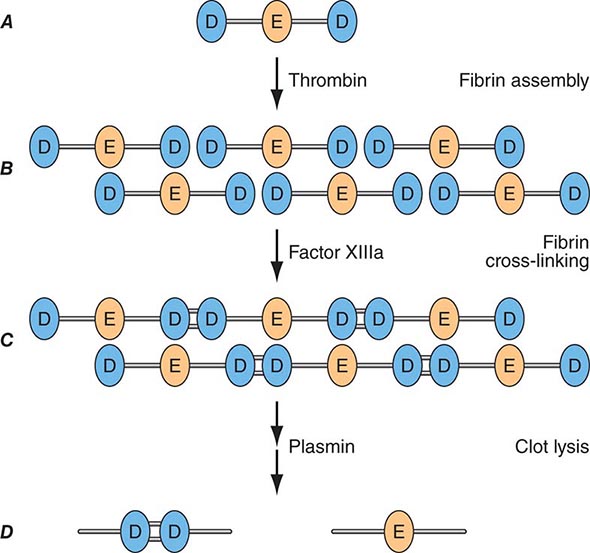
FIGURE 78-2 Fibrin formation and dissolution. (A) Fibrinogen is a trinodular structure consisting of two D domains and one E domain. Thrombin activation results in an ordered lateral assembly of protofibrils (B) with noncovalent associations. Factor XIIIa cross-links the D domains on adjacent molecules (C). Fibrin and fibrinogen (not shown) lysis by plasmin occurs at discrete sites and results in intermediary fibrin(ogen) degradation products (not shown). D-Dimers are the product of complete lysis of fibrin (D), maintaining the cross-linked D domains.
The assembly of the clotting factors on activated cell membrane surfaces greatly accelerates their reaction rates and also serves to localize blood clotting to sites of vascular injury. The critical cell membrane components, acidic phospholipids, are not normally exposed on resting cell membrane surfaces. However, when platelets, monocytes, and endothelial cells are activated by vascular injury or inflammatory stimuli, the procoagulant head groups of the membrane anionic phospholipids become translocated to the surfaces of these cells or released as part of microparticles, making them available to support and promote the plasma coagulation reactions.
ANTITHROMBOTIC MECHANISMS
Several physiologic antithrombotic mechanisms act in concert to prevent clotting under normal circumstances. These mechanisms operate to preserve blood fluidity and to limit blood clotting to specific focal sites of vascular injury. Endothelial cells have many antithrombotic effects. They produce prostacyclin, nitric oxide, and ectoADPase/CD39, which act to inhibit platelet binding, secretion, and aggregation. Endothelial cells produce anticoagulant factors including heparan proteoglycans, antithrombin, TF pathway inhibitor, and thrombomodulin. They also activate fibrinolytic mechanisms through the production of tissue plasminogen activator 1, urokinase, plasminogen activator inhibitor, and annexin-2. The sites of action of the major physiologic antithrombotic pathways are shown in Fig. 78-3.
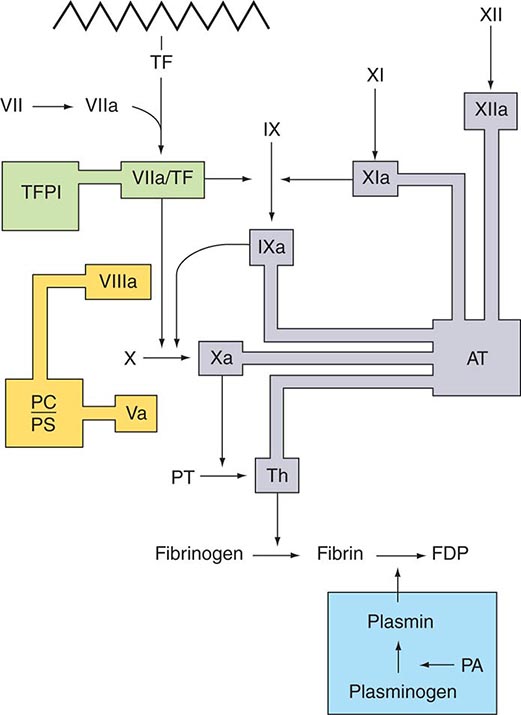
FIGURE 78-3 Sites of action of the four major physiologic antithrombotic pathways: antithrombin (AT); protein C/S (PC/PS); tissue factor pathway inhibitor (TFPI); and the fibrinolytic system, consisting of plasminogen, plasminogen activator (PA), and plasmin. PT, prothrombin; Th, thrombin; FDP, fibrin(ogen) degradation products. (Modified from BA Konkle, AI Schafer, in DP Zipes et al [eds]: Braunwald’s Heart Disease, 7th ed. Philadelphia, Saunders, 2005.)
Antithrombin (or antithrombin III) is the major plasma protease inhibitor of thrombin and the other clotting factors in coagulation. Antithrombin neutralizes thrombin and other activated coagulation factors by forming a complex between the active site of the enzyme and the reactive center of antithrombin. The rate of formation of these inactivating complexes increases by a factor of several thousand in the presence of heparin. Antithrombin inactivation of thrombin and other activated clotting factors occurs physiologically on vascular surfaces, where glycosoaminoglycans, including heparan sulfates, are present to catalyze these reactions. Inherited quantitative or qualitative deficiencies of antithrombin lead to a lifelong predisposition to venous thromboembolism.
Protein C is a plasma glycoprotein that becomes an anticoagulant when it is activated by thrombin. The thrombin-induced activation of protein C occurs physiologically on thrombomodulin, a transmembrane proteoglycan-binding site for thrombin on endothelial cell surfaces. The binding of protein C to its receptor on endothelial cells places it in proximity to the thrombin-thrombomodulin complex, thereby enhancing its activation efficiency. Activated protein C acts as an anticoagulant by cleaving and inactivating activated factors V and VIII. This reaction is accelerated by a cofactor, protein S, which, like protein C, is a glycoprotein that undergoes vitamin K–dependent posttranslational modification. Quantitative or qualitative deficiencies of protein C or protein S, or resistance to the action of activated protein C by a specific mutation at its target cleavage site in factor Va (factor V Leiden), lead to hypercoagulable states.
Tissue factor pathway inhibitor (TFPI) is a plasma protease inhibitor that regulates the TF-induced extrinsic pathway of coagulation. TFPI inhibits the TF/factor VIIa/factor Xa complex, essentially turning off the TF/factor VIIa initiation of coagulation, which then becomes dependent on the “amplification loop” via factor XI and factor VIII activation by thrombin. TFPI is bound to lipoprotein and can also be released by heparin from endothelial cells, where it is bound to glycosaminoglycans, and from platelets. The heparin-mediated release of TFPI may play a role in the anticoagulant effects of unfractionated and low-molecular-weight heparins.
THE FIBRINOLYTIC SYSTEM
Any thrombin that escapes the inhibitory effects of the physiologic anticoagulant systems is available to convert fibrinogen to fibrin. In response, the endogenous fibrinolytic system is then activated to dispose of intravascular fibrin and thereby maintain or reestablish the patency of the circulation. Just as thrombin is the key protease enzyme of the coagulation system, plasmin is the major protease enzyme of the fibrinolytic system, acting to digest fibrin to fibrin degradation products. The general scheme of fibrinolysis and its control is shown in Fig. 78-4.
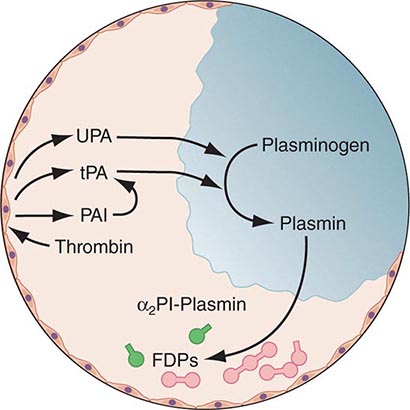
FIGURE 78-4 A schematic diagram of the fibrinolytic system. Tissue plasminogen activator (tPA) is released from endothelial cells, binds the fibrin clot, and activates plasminogen to plasmin. Excess fibrin is degraded by plasmin to distinct degradation products (FDPs). Any free plasmin is complexed with α2-antiplasmin (α2Pl). PAI, plasminogen activator inhibitor; UPA, urokinase-type plasminogen activator.
The plasminogen activators, tissue type plasminogen activator (tPA) and the urokinase-type plasminogen activator (uPA), cleave the Arg560-Val561 bond of plasminogen to generate the active enzyme plasmin. The lysine-binding sites of plasmin (and plasminogen) permit it to bind to fibrin, so that physiologic fibrinolysis is “fibrin specific.” Both plasminogen (through its lysine-binding sites) and tPA possess specific affinity for fibrin and thereby bind selectively to clots. The assembly of a ternary complex, consisting of fibrin, plasminogen, and tPA, promotes the localized interaction between plasminogen and tPA and greatly accelerates the rate of plasminogen activation to plasmin. Moreover, partial degradation of fibrin by plasmin exposes new plasminogen and tPA-binding sites in carboxy-terminus lysine residues of fibrin fragments to enhance these reactions further. This creates a highly efficient mechanism to generate plasmin focally on the fibrin clot, which then becomes plasmin’s substrate for digestion to fibrin degradation products.
Plasmin cleaves fibrin at distinct sites of the fibrin molecule, leading to the generation of characteristic fibrin fragments during the process of fibrinolysis (Fig. 78-2). The sites of plasmin cleavage of fibrin are the same as those in fibrinogen. However, when plasmin acts on covalently cross-linked fibrin, D-dimers are released; hence, D-dimers can be measured in plasma as a relatively specific test of fibrin (rather than fibrinogen) degradation. D-Dimer assays can be used as sensitive markers of blood clot formation and have been validated for clinical use to exclude the diagnosis of deep venous thrombosis (DVT) and pulmonary embolism in selected populations. In addition, D-dimer measurement can be used to stratify patients, particularly women, for risk of recurrent venous thromboembolism (VTE) when measured 1 month after discontinuation of anticoagulation given for treatment of an initial idiopathic event. D-Dimer levels may be elevated in the absence of VTE in elderly people.
Physiologic regulation of fibrinolysis occurs primarily at three levels: (1) plasminogen activator inhibitors (PAIs), specifically PAI-1 and PAI-2, inhibit the physiologic plasminogen activators; (2) the thrombin-activatable fibrinolysis inhibitor (TAFI) limits fibrinolysis; and (3) α2-antiplasmin inhibits plasmin. PAI-1 is the primary inhibitor of tPA and uPA in plasma. TAFI cleaves the N-terminal lysine residues of fibrin, which aid in localization of plasmin activity. α2-Antiplasmin is the main inhibitor of plasmin in human plasma, inactivating any nonfibrin clot-associated plasmin.



