Asthma
KEY CONCEPTS
![]() Asthma is a disease of increasing prevalence that is a result of genetic predisposition and environmental interactions; it is one of the most common chronic diseases of childhood.
Asthma is a disease of increasing prevalence that is a result of genetic predisposition and environmental interactions; it is one of the most common chronic diseases of childhood.
![]() Asthma is primarily a chronic inflammatory disease of the airways of the lung for which there is no known cure or primary prevention; the immunohistopathologic features include cell infiltration by neutrophils, eosinophils, T-helper type 2 lymphocytes, mast cells, and epithelial cells.
Asthma is primarily a chronic inflammatory disease of the airways of the lung for which there is no known cure or primary prevention; the immunohistopathologic features include cell infiltration by neutrophils, eosinophils, T-helper type 2 lymphocytes, mast cells, and epithelial cells.
![]() Asthma is characterized by either the intermittent or persistent presence of highly variable degrees of airflow obstruction from airway wall inflammation and bronchial smooth muscle constriction; in some patients, persistent changes in airway structure occur.
Asthma is characterized by either the intermittent or persistent presence of highly variable degrees of airflow obstruction from airway wall inflammation and bronchial smooth muscle constriction; in some patients, persistent changes in airway structure occur.
![]() The inflammatory process in asthma is treated most effectively with corticosteroids, with the inhaled corticosteroids having the greatest efficacy and safety profile for long-term management.
The inflammatory process in asthma is treated most effectively with corticosteroids, with the inhaled corticosteroids having the greatest efficacy and safety profile for long-term management.
![]() Bronchial smooth muscle constriction is prevented or treated most effectively with inhaled β2-adrenergic receptor agonists.
Bronchial smooth muscle constriction is prevented or treated most effectively with inhaled β2-adrenergic receptor agonists.
![]() Variability in response to medications requires individualization of therapy within existing evidence-based guidelines for management. This is most evident in patients with severe asthma phenotypes.
Variability in response to medications requires individualization of therapy within existing evidence-based guidelines for management. This is most evident in patients with severe asthma phenotypes.
![]() Ongoing patient education, for a partnership in asthma care, is essential for optimal patient outcomes and includes trigger avoidance and self-management techniques.
Ongoing patient education, for a partnership in asthma care, is essential for optimal patient outcomes and includes trigger avoidance and self-management techniques.
Asthma has been known since antiquity, yet it is a disease that still defies precise definition. The word asthma is of Greek origin and means “panting.” More than 2,000 years ago, Hippocrates used the word asthma to describe episodic shortness of breath; however, the first detailed clinical description of the asthmatic patient was made by Aretaeus in the second century.1 The National Institutes of Health, National Asthma Education and Prevention Program (NAEPP) Expert Panel Report 3 (EPR3), has provided the following working definition of asthma2:
Asthma is a chronic inflammatory disorder of the airways in which many cells and cellular elements play a role: in particular, mast cells, eosinophils, T-lymphocytes, macrophages, neutrophils, and epithelial cells. In susceptible individuals, this inflammation causes recurrent episodes of wheezing, breathlessness, chest tightness, and coughing, particularly at night or in the early morning. These episodes are usually associated with widespread but variable airflow obstruction that is often reversible either spontaneously or with treatment. The inflammation also causes an associated increase in the existing bronchial hyperresponsiveness (BHR) to a variety of stimuli. Reversibility of airflow limitation may be incomplete in some patients with asthma.
This definition encompasses the important heterogeneity of the clinical presentation of asthma by describing the scientific and clinically accepted characteristics of asthma.
EPIDEMIOLOGY
![]() An estimated 25.7 million persons in the United States have asthma (about 8.4% of the population).3 Asthma is the most common chronic disease among children in the United States, with approximately 7 million children affected. The prevalence rate is highest in children 0 to 17 years of age at 9.5%.3 In the United States, as in other industrialized countries, the prevalence of asthma is increasing from 7.3% in 2001. Asthma prevalence is higher in persons with incomes below 100% of poverty level at 11.2% and in blacks 11.2% and multiple races 14.1%. Asthma accounts for 1.6% of all ambulatory care visits (10.6 million physician office visits and 1.2 million hospital outpatient visits) and resulted in 440,000 hospitalizations and 1.7 million emergency department (ED) visits in 2006 (both declined from peaks in the 1990s).4 It is the third leading cause of preventable hospitalization in the United States; however, hospitalizations have decreased per 100 patients with asthma since 2001. Asthma accounts for more than 12.8 million missed school days per year.4 In young children (0 to 10 years of age), the risk of asthma is greater in boys than in girls, becomes about equal during puberty, and then is greater in women than in men.4
An estimated 25.7 million persons in the United States have asthma (about 8.4% of the population).3 Asthma is the most common chronic disease among children in the United States, with approximately 7 million children affected. The prevalence rate is highest in children 0 to 17 years of age at 9.5%.3 In the United States, as in other industrialized countries, the prevalence of asthma is increasing from 7.3% in 2001. Asthma prevalence is higher in persons with incomes below 100% of poverty level at 11.2% and in blacks 11.2% and multiple races 14.1%. Asthma accounts for 1.6% of all ambulatory care visits (10.6 million physician office visits and 1.2 million hospital outpatient visits) and resulted in 440,000 hospitalizations and 1.7 million emergency department (ED) visits in 2006 (both declined from peaks in the 1990s).4 It is the third leading cause of preventable hospitalization in the United States; however, hospitalizations have decreased per 100 patients with asthma since 2001. Asthma accounts for more than 12.8 million missed school days per year.4 In young children (0 to 10 years of age), the risk of asthma is greater in boys than in girls, becomes about equal during puberty, and then is greater in women than in men.4
Ethnic minorities continue to share the burden of asthma disproportionately. African Americans are two times as likely to be hospitalized and approximately two times more likely to die from asthma than whites.3 Hispanics in general, with the exception of Puerto Ricans, at 16.1% prevalence have lower disease and hospitalization rates than African Americans or whites.
The estimated direct medical cost of asthma in the United States in 2007 was $14.7 billion.4 The societal burden of asthma (indirect medical expenditures: loss of productivity and death) in the United States was $5 billion. Prescription drugs were the largest single direct medical expenditure at $6.2 billion; however, the combined costs of emergency care of acute asthma exacerbations make up 36% of direct medical costs.4
The natural history of asthma is still not well defined. Although asthma can occur at any time, it is principally a pediatric disease, with most patients being diagnosed by 5 years of age and up to 50% of children having symptoms by 2 years of age.2 Between 30% and 70% of children with asthma will improve markedly or become symptom-free by early adulthood; chronic disease persists in about 30% to 40% of patients, and generally 20% or less develop severe chronic disease.2 Predictors of persistent adult asthma include atopy, onset during school age, and presence of bronchial hyperresponsiveness (BHR).2 Diminished lung growth may occur in some children (approximately 10%) with asthma.2
In adults, most longitudinal studies have suggested a more rapid rate of decline in lung function in asthmatics than in nonasthmatic normals, primarily reflected in forced expiratory volume in 1 second (FEV1).2 However, the annual decline in FEV1 is less than in smokers or in patients with a diagnosis of emphysema. In general, individuals with less frequent asthma attacks and normal lung function on initial assessment have higher remission rates, whereas smokers have the lowest remission and highest relapse rates.2 The level of BHR tends to predict the rate of decline in FEV1, with a greater decline with high levels of BHR.2 Thus, airway obstruction in asthma may become irreversible and also worsen over time owing to airway remodeling (see below).2 However, most patients do not die from long-term progression of their disease and their life span is not different from the general population.2
As with prevalence and morbidity, mortality from acute exacerbations of asthma has been decreasing over the past 10 years, with a death rate of 0.14 per 1,000 persons with asthma reported in 2009.3 Despite the relatively low number of asthma deaths, 80% to 90% are preventable.2 Most deaths from asthma occur outside the hospital, and death is rare after hospitalization. The most common cause of death from asthma is inadequate assessment of the severity of airway obstruction by the patient or physician and inadequate therapy. The most common cause of death in hospitalized patients is also inadequate or inappropriate therapy. Thus, the key to prevention of death from asthma, as advocated by the U.S. NAEPP, is education.2
ETIOLOGY
![]() Epidemiologic studies strongly support the concept of a genetic predisposition plus environmental interaction to the development of asthma, yet the picture remains complex and incomplete.5 Genetic factors account for 60% to 80% of the susceptibility. Asthma represents a complex genetic disorder in that the asthma phenotype is likely a result of polygenic inheritance or different combinations of genes. Initial searches focused on establishing links between atopy (genetically determined state of hypersensitivity to environmental allergens) and asthma, but more recent genome-wide searches have found linkages with genes for metalloproteinases (e.g., ADAM33) and handling bacteria (CHI3L1).5 Although genetic predisposition to atopy is a significant risk factor for developing asthma, not all atopic individuals develop asthma, nor do all patients with asthma exhibit atopy. Disparate phenotypes of asthma (progressive or remodeled vs. nonprogressive) are likely genetically determined.5
Epidemiologic studies strongly support the concept of a genetic predisposition plus environmental interaction to the development of asthma, yet the picture remains complex and incomplete.5 Genetic factors account for 60% to 80% of the susceptibility. Asthma represents a complex genetic disorder in that the asthma phenotype is likely a result of polygenic inheritance or different combinations of genes. Initial searches focused on establishing links between atopy (genetically determined state of hypersensitivity to environmental allergens) and asthma, but more recent genome-wide searches have found linkages with genes for metalloproteinases (e.g., ADAM33) and handling bacteria (CHI3L1).5 Although genetic predisposition to atopy is a significant risk factor for developing asthma, not all atopic individuals develop asthma, nor do all patients with asthma exhibit atopy. Disparate phenotypes of asthma (progressive or remodeled vs. nonprogressive) are likely genetically determined.5
![]() Environmental risk factors for the development of asthma include socioeconomic status, family size, exposure to secondhand tobacco smoke in infancy and in utero, allergen exposure, urbanization, respiratory syncytial virus (RSV) infection, and decreased exposure to common childhood infectious agents.6,7 The “hygiene hypothesis” proposes that genetically susceptible individuals develop allergies and asthma by allowing the allergic immunologic system (T-helper cell type 2 [Th2] lymphocytes) to develop instead of the system to fight infections (T-helper type 1 [Th1] lymphocytes) and may explain the increase of asthma in developed countries.6,7 The first 2 years of life appear to be most important for the exposures to produce an alteration in the immune response system.6 The hygiene hypothesis is supported by studies demonstrating a lower risk for asthma in children who are exposed to high levels of bacteria or endotoxin, in those with a large number of older siblings, in those with early enrollment into child care, in those with exposure to cats and dogs early in life, or in those with exposure to fewer antibiotics.5–7
Environmental risk factors for the development of asthma include socioeconomic status, family size, exposure to secondhand tobacco smoke in infancy and in utero, allergen exposure, urbanization, respiratory syncytial virus (RSV) infection, and decreased exposure to common childhood infectious agents.6,7 The “hygiene hypothesis” proposes that genetically susceptible individuals develop allergies and asthma by allowing the allergic immunologic system (T-helper cell type 2 [Th2] lymphocytes) to develop instead of the system to fight infections (T-helper type 1 [Th1] lymphocytes) and may explain the increase of asthma in developed countries.6,7 The first 2 years of life appear to be most important for the exposures to produce an alteration in the immune response system.6 The hygiene hypothesis is supported by studies demonstrating a lower risk for asthma in children who are exposed to high levels of bacteria or endotoxin, in those with a large number of older siblings, in those with early enrollment into child care, in those with exposure to cats and dogs early in life, or in those with exposure to fewer antibiotics.5–7
Risk factors for early (<3 years of age) recurrent wheezing associated with viral infections include low birth weight, male gender, and parental smoking. However, this early pattern is due to smaller airways, and these risk factors are not necessarily risk factors for asthma in later life.6 Atopy is the predominant risk factor for children to have continued asthma.6,7 Asthma can begin in adults later in life. Occupational asthma in previously healthy individuals emphasizes the effect of environment on the development of asthma.8 The heterogeneity of the asthma phenotype appears most obvious when listing the diverse triggers of bronchospasm2,6 (Table 15-1). The various triggers have relative degrees of importance from patient to patient. Environmental exposures are the most important precipitants of severe asthma exacerbations9 (see Table 15-1). Epidemics of severe asthma in cities have followed exposures to high concentrations of aeroallergens.9 Viral respiratory tract infections remain the single most significant precipitant of severe asthma in children and are an important trigger in adults as well.10 Other possible factors include air pollution, sinusitis, food preservatives, and drugs.
TABLE 15-1 List of Agents and Events Triggering Asthma

PATHOPHYSIOLOGY
![]() The major characteristics of asthma include a variable degree of airflow obstruction (related to bronchospasm, edema, and mucus hypersecretion), BHR, and airway inflammation (Fig. 15-1). To understand the pathogenetic mechanisms that underlie the many phenotypes of asthma, it is critical to identify factors that initiate, intensify, and modulate the inflammatory response of the airways and to determine how these processes produce the characteristic airway abnormalities.
The major characteristics of asthma include a variable degree of airflow obstruction (related to bronchospasm, edema, and mucus hypersecretion), BHR, and airway inflammation (Fig. 15-1). To understand the pathogenetic mechanisms that underlie the many phenotypes of asthma, it is critical to identify factors that initiate, intensify, and modulate the inflammatory response of the airways and to determine how these processes produce the characteristic airway abnormalities.
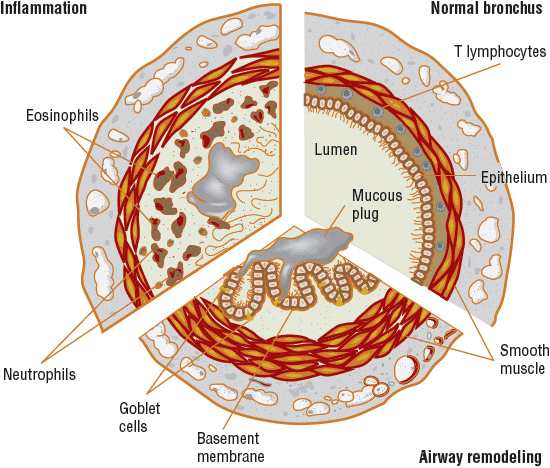
FIGURE 15-1 Representative illustration of the pathology found in the asthmatic bronchus compared with a normal bronchus (upper right). Each section demonstrates how the lumen is narrowed. Hypertrophy of the basement membrane, mucus plugging, smooth muscle hypertrophy, and constriction contribute (lower section). Inflammatory cells infiltrate, producing submucosal edema, and epithelial desquamation fills the airway lumen with cellular debris and exposes the airway smooth muscle to other mediators (upper left).
Acute Inflammation
Inhaled allergen challenge models contribute most to our understanding of acute inflammation in asthma.7 Inhaled allergen challenge in allergic patients leads to an early phase reaction that, in some cases, may be followed by a late-phase reaction. The activation of cells bearing allergen-specific immunoglobulin E (IgE) initiates the early phase reaction. It is characterized by the rapid activation of airway mast cells and macrophages leading to the rapid release of proinflammatory mediators such as histamine, eicosanoids, and reactive oxygen (O2) species that induce contraction of airway smooth muscle, mucus secretion, and vasodilation.7 The bronchial microcirculation has an essential role in this inflammatory process. Inflammatory mediators induce microvascular leakage with exudation of plasma in the airways.7 Acute plasma protein leakage induces a thickened, engorged, and edematous airway wall and a consequent narrowing of the airway lumen. Plasma exudation may compromise epithelial integrity, and the presence of plasma in the lumen may reduce mucus clearance.7 Plasma proteins also may promote the formation of exudative plugs mixed with mucus and inflammatory and epithelial cells. Together these effects contribute to airflow obstruction (Fig. 15-1).
The late-phase inflammatory reaction occurs 6 to 9 hours after allergen provocation and involves the recruitment and activation of eosinophils, CD4+ thymically derived lymphocytes (T cells), basophils, neutrophils, and macrophages.7 There is selective retention of airway T cells, the expression of adhesion molecules, and the release of selected proinflammatory mediators and cytokines involved in the recruitment and activation of inflammatory cells.7 The activation of T cells after allergen challenge leads to the release of Th2-like cytokines that may modulate the late-phase response.7 The release of preformed cytokines by mast cells is the likely initial trigger for the early recruitment of inflammatory cells that then recruit and induce the more persistent involvement by T cells.7 The enhancement of nonspecific BHR usually can be demonstrated after the late-phase reaction but not after the early phase reaction following allergen or occupational challenge.
Chronic Inflammation
Airway inflammation has been demonstrated in all forms of asthma, and an association between the extent of inflammation and the clinical severity of asthma has been demonstrated in selected studies.7 It is accepted that both central and peripheral airways are inflamed.
In asthma, all cells of the airways are involved and become activated (Fig. 15-2). Included are eosinophils, T cells, mast cells, macrophages, epithelial cells, fibroblasts, and bronchial smooth muscle cells. These cells also regulate airway inflammation and initiate the process of remodeling by the release of cytokines and growth factors.7,11
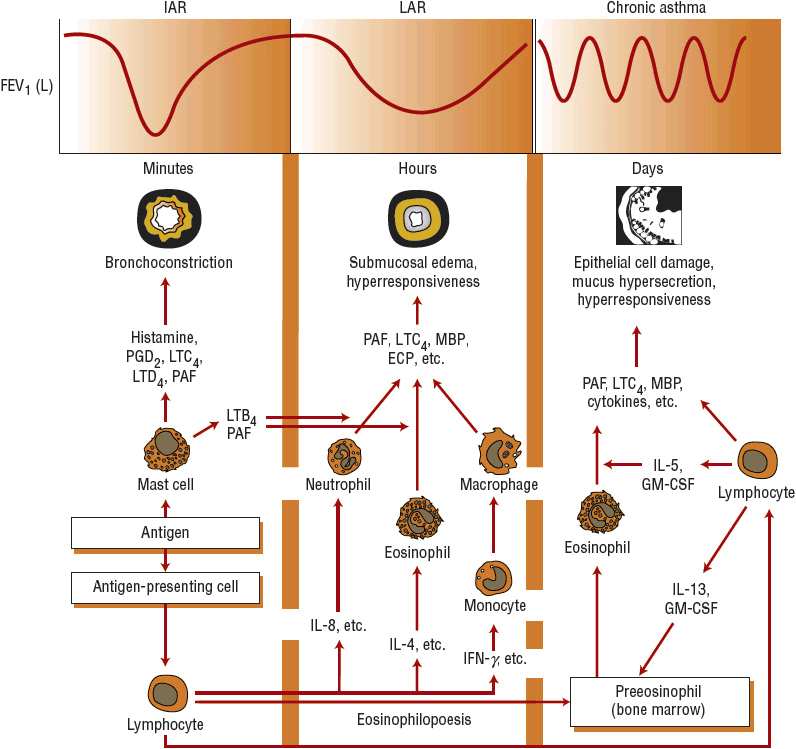
FIGURE 15-2 Diagrammatic presentation of the relationship between inflammatory cells, lipid and preformed mediators, inflammatory cytokines, and proposed pathogenesis and clinical presentation in asthma. See text for details. (GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; LT, leukotriene; MBP, major basic protein; PAF, platelet-activating factor; PG, prostaglandin.)
Epithelial Cells
Bronchial epithelial cells participate in mucociliary clearance and removal of noxious agents; however, they also enhance inflammation by releasing eicosanoids, peptidases, matrix proteins, cytokines, chemokines, and nitric oxide (NO).7,11 Epithelial cells can be activated by IgE-dependent mechanisms, viruses, pollutants, or histamine. In asthma, especially fatal asthma, extensive epithelial shedding occurs. The functional consequences of epithelial shedding may include heightened airway responsiveness, release of the chemokine eotaxin that attracts eosinophils, altered permeability of the airway mucosa, depletion of epithelial-derived relaxant factors, and loss of enzymes responsible for degrading proinflammatory neuropeptides. The integrity of airway epithelium may influence the sensitivity of the airways to various provocative stimuli. Epithelial cells also may be important in the regulation of airway remodeling and fibrosis.7,11
Eosinophils
Eosinophils play an effector role in asthma by releasing proinflammatory mediators, cytotoxic mediators, and cytokines.7 Circulating eosinophils migrate to the airways by cell rolling, through interactions with selectins, and eventually adhere to the endothelium through the binding of integrins to adhesion proteins (vascular cell adhesion molecule 1 [VCAM-1] and intercellular adhesion molecule 1 [ICAM-1]). As eosinophils enter the matrix of the membrane, their survival is prolonged by interleukin 5 (IL-5) and granulocyte-macrophage colony-stimulating factor (GM-CSF). On activation, eosinophils release inflammatory mediators such as leukotrienes (LTs) and granule proteins to injure airway tissue.7
Lymphocytes
Mucosal biopsy specimens from patients with asthma contain lymphocytes, many of which express surface markers of inflammation. There are two types of T-helper CD4+ cells. Th1 cells produce IL-2 and interferon-γ (IFN-γ), both essential for cellular defense mechanisms. Th2 cells produce cytokines (IL-4, -5, and -13) that mediate allergic inflammation. It is known that Th1 cytokines inhibit the production of Th2 cytokines, and vice versa. It is hypothesized that allergic asthmatic inflammation results from a Th2-mediated mechanism (an imbalance between Th1 and Th2 cells).7 However, more recently it has been observed that there exists a low Th2 cytokine phenotype of asthma in adults that appears more resistant to usual therapies for asthma.12
Th1 and Th2 Cell Imbalance
The T-cell population in the cord blood of newborn infants is skewed toward a Th2 phenotype.6,7 The extent of the imbalance between Th1 and Th2 cells (as indicated by diminished IFN-γ production) during the neonatal phase may predict the subsequent development of allergic disease, asthma, or both. It has been suggested that infants at high risk of asthma and allergies should be exposed to stimuli that upregulate Th1-mediated responses in order to restore the balance during a critical time in the development of the immune system and the lungs.6
The basic premise of the hygiene hypothesis is that the newborn’s immune system needs timely and appropriate environmental stimuli to create a balanced immune response. Factors that enhance Th1-mediated responses include infection with Mycobacterium tuberculosis, measles virus, and hepatitis A virus; endotoxin exposure; increased exposure to infections through contact with older siblings; and daycare attendance during the first 6 months of life. Restoration of the balance between Th1 and Th2 cells may be impeded by frequent administration of oral antibiotics, with concomitant alterations in GI flora. Other factors favoring the Th2 phenotype include residence in an industrialized country, urban environment exposure, diet, and sensitization to house dust mites and cockroaches.6 Immune “imprinting” may begin in utero by transplacental transfer of allergens and cytokines.
Mast Cell
Mast cell degranulation is important in the initiation of immediate responses following exposure to allergens.2 Mast cells reside throughout the walls of the respiratory tract, and increased numbers of these cells (threefold to fivefold) have been described in the airways of allergic asthmatics.7 Once binding of allergen to cell-bound IgE occurs, mediators such as histamine; eosinophil and neutrophil chemotactic factors; LTs C4, D4, and E4; prostaglandins; platelet-activating factor (PAF); and others are released from mast cells (see Fig. 15-2). Histologic examination has revealed decreased numbers of granulated mast cells in the airways of patients who have died from acute asthma attacks, suggesting that mast cell degranulation is a contributing factor. Sensitized mast cells are also activated by osmotic stimuli to account for exercise-induced bronchospasm (EIB).13
Alveolar Macrophages
The primary function of alveolar macrophages in the normal airway is to serve as “scavengers,” engulfing and digesting bacteria and other foreign materials. Macrophages are found in large and small airways, ideally located for affecting the asthmatic response. A number of mediators produced and released by macrophages have been identified, including PAF, LTB4, LTC4, and LTD4.7 Additionally, alveolar macrophages are able to produce neutrophil chemotactic factor and eosinophil chemotactic factor, which in turn amplify the inflammatory process.
Neutrophils
The role of neutrophils in the pathogenesis of asthma remains somewhat unclear because they normally may be present in the airways and usually do not infiltrate tissues showing chronic allergic inflammation despite the potential to participate in late-phase inflammatory reactions. However, high numbers of neutrophils have been observed in the airways of patients who died from sudden-onset fatal asthma and in those with severe disease.14 This suggests that neutrophils may play a pivotal role in the disease process, at least in some patients with long-standing or corticosteroid-resistant asthma.14 The neutrophil also can be a source for a variety of mediators, including PAF, prostaglandins, thromboxanes, and LTs, that contribute to BHR and airway inflammation.14
Fibroblasts and Myofibroblasts
Fibroblasts are found frequently in connective tissue. Human lung fibroblasts may behave as inflammatory cells on activation by IL-4 and IL-13. The myofibroblast may contribute to the regulation of inflammation via the release of cytokines and to tissue remodeling. In asthma, myofibroblasts are increased in numbers beneath the reticular basement membrane, and there is an association between their numbers and the thickness of the reticular basement membrane.7,11
Inflammatory Mediators
Associated with asthma for many years, histamine is capable of inducing smooth muscle constriction and bronchospasm and is thought to play a role in mucosal edema and mucus secretion.2 Lung mast cells are an important source of histamine. The release of histamine can be stimulated by exposure of the airways to a variety of factors, including physical stimuli (airway drying with exercise) and relevant allergens.7 Histamine is involved in acute bronchospasm following allergen exposure; however, other mediators such as LTs are also involved.
Besides histamine release, mast cell degranulation releases ILs, proteases, and other enzymes that activate the production of other mediators of inflammation. Several classes of important mediators, including arachidonic acid and its metabolites (i.e., prostaglandins, LTs, and PAF), are derived from cell membrane phospholipids.
Once arachidonic acid is released, it can be metabolized by the enzyme cyclooxygenase to form prostaglandins. Prostaglandin D2 is a potent bronchoconstricting agent; however, it is unlikely to produce sustained effects and its role in asthma remains to be determined. Similarly, prostaglandin F2α is a potent bronchoconstrictor in patients with asthma and can enhance the effects of histamine.2,7 However, its pathophysiologic role in asthma is unclear. Another cyclooxygenase product, prostacyclin (prostaglandin I2), is known to be produced in the lung and may contribute to inflammation and edema owing to its effects as a vasodilator.
Thromboxane A2 is produced by alveolar macrophages, fibroblasts, epithelial cells, neutrophils, and platelets within the lung.7 It may have several effects, including bronchoconstriction, involvement in the late asthmatic response, and involvement in the development of airway inflammation and BHR.
The 5-lipoxygenase pathway of arachidonic acid metabolism is responsible for the production of the cysteinyl LTs.7 LTC4, LTD4, and LTE4 are released during inflammatory processes in the lung. LTs D4 and E4 share a common receptor (LTD4 receptor) that, when stimulated, produces bronchospasm, mucus secretion, microvascular permeability, and airway edema, whereas LTB4 is involved with granulocyte chemotaxis.
Thought to be produced by macrophages, eosinophils, and neutrophils within the lung, PAF is involved in the mediation of bronchospasm, sustained induction of BHR, edema formation, and chemotaxis of eosinophils.7
Adhesion Molecules
Adhesion molecules are glycoproteins that facilitate infiltration and migration of inflammatory cells to the site of inflammation. They have additional functions involved in the inflammatory process aside from promoting cell adhesion, including activation of cells and cell–cell communication, and promoting cellular migration and infiltration.2 The many adhesion molecules are divided into families on the basis of their chemical structure. These families are the integrins, cadherins, immunoglobulin supergene family, selectins, vascular adressins, and carbohydrate ligands.7 Those thought to be important in inflammation include the integrins, immunoglobulin supergene family, selectins, and carbohydrate ligands, including ICAM-1 and VCAM-1.7 Adhesion molecules are found on a variety of cells, such as neutrophils, monocytes, lymphocytes, basophils, eosinophils, granulocytes, platelets, endothelial cells, and epithelial cells, and can be expressed or activated by the many inflammatory mediators present in asthma.7
Clinical Consequences of Chronic Inflammation
Chronic inflammation is associated with nonspecific BHR and increases the risk of asthma exacerbations. Exacerbations are characterized by increased symptoms and worsening airway obstruction over a period of days or even weeks, and rarely hours. Hyperresponsiveness of the airways to physical, chemical, and pharmacologic stimuli is a hallmark of asthma.2 BHR also occurs in some patients with chronic bronchitis and allergic rhinitis.2 Normal healthy subjects also may develop a transient BHR after viral respiratory infections or ozone exposure. However, the degree of BHR in patients with asthma is quantitatively greater than in other populations. Bronchial responsiveness of the general population fits a unimodal distribution that is skewed toward increased reactivity; individuals with clinical asthma represent the extreme end of this distribution. The degree of BHR within asthma correlates with its clinical course and medication requirement necessary to control symptoms.2 Patients with mild symptoms or in remission demonstrate lower levels of BHR.
The current understanding is that the BHR seen in asthma is at least in part due to and correlative with the extent of airway inflammation.2 Airway remodeling also correlates somewhat with BHR.11
Remodeling of the Airways
Acute inflammation is a beneficial, nonspecific response of tissues to injury and generally leads to repair and restoration of the normal structure and function. In contrast, asthma represents a chronic inflammatory process of the airways followed by healing that in some may result in altered structure referred to as remodeling.11 Repair involves replacement of injured tissue by parenchymal cells of the same type and replacement by connective tissue and its maturation into scar tissue. In asthma, remodeling presents as extracellular matrix fibrosis, an increase in smooth muscle and mucus gland mass, and angiogenesis.11
The precise mechanisms of remodeling of the airways are under intense study. Airway remodeling is of concern because it may represent an irreversible process that can have more serious sequelae such as the development of chronic obstructive pulmonary disease (COPD).2,11 Observations in children with asthma indicate that some loss of lung function may occur during the first 5 years of life.6 Importantly, no current therapies have been shown to alter either early decreased lung growth or later progressive loss of lung function.
Mucus Production
The mucociliary system is the lung’s primary defense mechanism against irritants and infectious agents. Mucus, composed of 95% water and 5% glycoproteins, is produced by bronchial epithelial glands and goblet cells.7 The lining of the airways consists of a continuous aqueous layer controlled by active ion transport across the epithelium in which water moves toward the lumen along the concentration gradient. Catecholamines and vagal stimulation enhance the ion transport and fluid movement. Mucus transport depends on its viscoelastic properties. Mucus that is either too watery or too viscous will not be transported optimally. The exudative inflammatory process and sloughing of epithelial cells into the airway lumen impair mucociliary transport. The bronchial glands are increased in size and the goblet cells are increased in size and number in asthma. Expectorated mucus from patients with asthma tends to have a high viscosity. The mucus plugs in the airways of patients who died in status asthmaticus are tenacious and tend to be connected by mucous strands to the goblet cells. Asthmatic airways also may become plugged with casts consisting of epithelial and inflammatory cells. Although it is tempting to speculate that death from asthma attacks is a result of the mucus plugging resulting in irreversible obstruction, there is no direct evidence for this. Autopsies of asthmatics who died from other causes have shown similar pathology. In addition, some patients who have died of sudden severe asthma did not show the characteristic mucus plugging on necropsy.7
Airway Smooth Muscle
The smooth muscle of the airways does not form a uniform coat around the airways but is wrapped around in a connecting network best described as a spiral arrangement.15 The muscle contraction displays a sphincteric action that is capable of completely occluding the airway lumen. The airway smooth muscle extends from the trachea through the respiratory bronchioles. When expressed as a percentage of wall thickness, the smooth muscle represents 5% of the large central airways and up to 20% of the wall thickness in the bronchioles. Total smooth muscle mass decreases rapidly past the terminal bronchioles to the alveoli, so the contribution of smooth muscle tone to airway diameter in this region is relatively small. In the large airways of asthmatics, smooth muscle may account for 11% of the wall thickness. It is possible that the increased smooth muscle mass of the asthmatic airways is important in magnifying and maintaining BHR in persistent disease. However, it appears that the hypertrophy and hyperplasia are secondary processes caused by chronic inflammation and are not the primary cause of BHR.15
Neural Control/Neurogenic Inflammation
The airway is innervated by parasympathetic, sympathetic, and nonadrenergic inhibitory nerves.2 Parasympathetic innervation of the smooth muscle consists of efferent motor fibers in the vagus nerves and sensory afferent fibers in the vagus and other nerves.15 The normal resting tone of human airway smooth muscle is maintained by vagal efferent activity. Maximum bronchoconstriction mediated by vagal stimulation occurs in the small bronchi and is absent in the small bronchioles. The nonmyelinated C fibers of the afferent system lie immediately beneath the tight junctions between epithelial cells lining the airway lumen.15 These endings probably represent the irritant receptors of the airways. Stimulation of these irritant receptors by mechanical stimulation, chemical and particulate irritants, and pharmacologic agents such as histamine produces reflex bronchoconstriction.7
The nonadrenergic, noncholinergic (NANC) nervous system has been described in the trachea and bronchi. Substance P, neurokinin A, neurokinin B, and vasoactive intestinal peptide (VIP) are the best characterized neurotransmitters in the NANC nervous system.7 VIP is an inhibitory neurotransmitter. Inflammatory cells in asthma can release peptidases that can degrade VIP, producing exaggerated reflex cholinergic bronchoconstriction. NANC excitatory neuropeptides such as substance P and neurokinin A are released by stimulation of C-fiber sensory nerve endings. The NANC system may play an important role in amplifying inflammation in asthma by releasing NO.
Nitric Oxide
NO is produced by cells within the respiratory tract. It has been thought to be a neurotransmitter of the NANC nervous system.16 Endogenous NO is generated from the amino acid L-arginine (L-Arg) by the enzyme NO synthase.16 There are three isoforms of NO synthase. One isoform is induced in response to proinflammatory cytokines, inducible NO synthase (iNOS), in airway epithelial cells and inflammatory cells of asthmatic airways.16 NO produces smooth muscle relaxation in the vasculature and bronchials; however, it appears to amplify the inflammatory process and is unlikely to be of therapeutic benefit. Investigations measuring the fraction of exhaled NO (FeNO) concentrations have suggested that it may be a useful measure of ongoing lower airway inflammation in patients with asthma and for guiding asthma therapy.16
CLINICAL PRESENTATION
Chronic Asthma
![]() Classic asthma is characterized by episodic dyspnea associated with wheezing; however, the clinical presentation of asthma is as diverse as the number of triggering events (see Clinical Presentation: Chronic Ambulatory Asthma below). Although wheezing is the characteristic symptom of asthma, the medical literature is replete with the warning that “not all that wheezes is asthma.” A wheeze is a high-pitched, whistling sound created by turbulent airflow through an obstructed airway, so any condition that produces significant obstruction can result in wheezing as a symptom. In addition, “all of asthma does not wheeze” is an equally justifiable warning. Patients may present with a chronic persistent cough as their only symptom.2
Classic asthma is characterized by episodic dyspnea associated with wheezing; however, the clinical presentation of asthma is as diverse as the number of triggering events (see Clinical Presentation: Chronic Ambulatory Asthma below). Although wheezing is the characteristic symptom of asthma, the medical literature is replete with the warning that “not all that wheezes is asthma.” A wheeze is a high-pitched, whistling sound created by turbulent airflow through an obstructed airway, so any condition that produces significant obstruction can result in wheezing as a symptom. In addition, “all of asthma does not wheeze” is an equally justifiable warning. Patients may present with a chronic persistent cough as their only symptom.2
CLINICAL PRESENTATION Chronic Ambulatory Asthma
There is no single diagnostic test for asthma. The diagnosis is based primarily on a good history2 (Table 15-2). The patient may have a family history of allergy or asthma or have symptoms of allergic rhinitis.2 Reversibility of airway obstruction following administration of a short-acting inhaled β2-agonist provides confirmation but is not by itself diagnostic. Patients with normal values of spirometry can be challenged by exercise or substances that produce bronchoconstriction, such as methacholine or mannitol, to determine if they have BHR, but, again, positive challenges are not diagnostic. Newer tests of inflammation in the airways such as induced sputum eosinophil and/or neutrophil counts and FeNO measurements are consistent with but not diagnostic of asthma.
TABLE 15-2 Sample Questionsa for the Diagnosis and Initial Assessment of Asthma

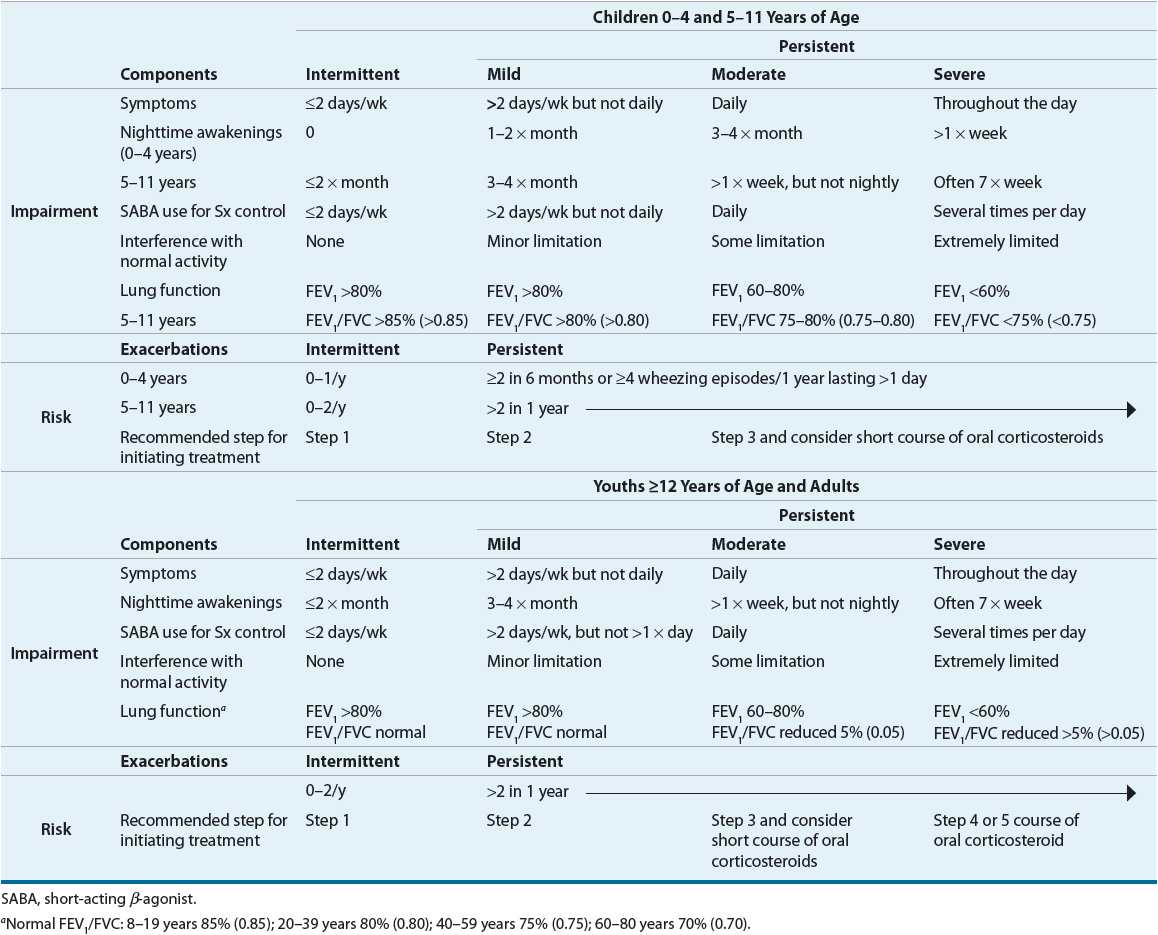
Asthma has a widely variable presentation from chronic daily symptoms to only intermittent symptoms. The intervals between symptoms can be days, weeks, months, or years. Asthma also can vary as to its severity, the intrinsic intensity of the disease process. Severity is most easily and directly measured in a patient who is not currently receiving asthma treatment. The NAEPP has provided a means of classifying asthma severity that is divided into two domains: impairment and risk.2 This classification system is individualized for three age groups (0 to 4, 5 to 11, and ≥12 years) and summarized in Table 15-3. The intermittent and/or chronic nature of symptoms does not necessarily determine the severity of symptoms during exacerbations. Asthma severity is determined by lung function, symptoms, nighttime awakenings, and interference with normal activity prior to therapy. Patients can present with a range from intermittent symptoms that require no medications or only occasional use of short-acting inhaled β2-agonists to severe persistent asthma symptoms despite treatment with multiple medications.
TABLE 15-3 Classifying Asthma Severity for Patients Who Are Not Currently Taking Long-Term Control Medications
Acute Severe Asthma
Uncontrolled asthma, with its inherent variability, can progress to an acute state where inflammation, airway edema, excessive mucus accumulation, and severe bronchospasm result in a profound airway narrowing that is poorly responsive to usual bronchodilator therapy2,9 (see Clinical Presentation: Acute Severe Asthma below). Although this progression is the most common scenario, some patients experience rapid-onset or hyperacute attacks.2,9 Hyperacute attacks are associated with neutrophilic as opposed to eosinophilic infiltration and resolve rapidly with bronchodilator therapy, suggesting that smooth muscle spasm is the major pathogenic mechanism.9,14 In most cases, ED visits for acute severe asthma represent the failure of an adequate therapeutic regimen to control persistent asthma. Underutilization of antiinflammatory drugs and excessive reliance on short-acting inhaled β2-agonists are the major risk factors for severe exacerbations.2 However, frequent exacerbations may represent a specific phenotype of asthma.9 A blunted perception of airway obstruction may predispose certain individuals to fatal asthma attacks.2
CLINICAL PRESENTATION Acute Severe Asthma
Exercise-Induced Bronchospasm
During vigorous exercise, pulmonary function measurements (FEV1 and PEF) in patients with asthma increase during the first few minutes but then begin to decrease after 6 to 8 minutes (Fig. 15-3).2 EIB is defined as a drop in FEV1 of 15% or greater from baseline (pre-exercise value).2 Most studies suggest that many patients with persistent asthma experience EIB.2 The exact pathogenesis of EIB is unknown, but heat loss and/or water loss from the central airways appears to play an important role.13 EIB is provoked more easily in cold, dry air; alternatively, warm, humid air can blunt or block it.13 Studies have demonstrated increased plasma histamine, cysteinyl LTs, prostaglandins, and tryptase concentrations during EIB, suggesting a role for mast cell degranulation.13 These findings have led to the development of inhaled mannitol, an osmotic agent, as an indirect pharmacologic bronchoprovocation test to assist in the diagnosis of asthma.17
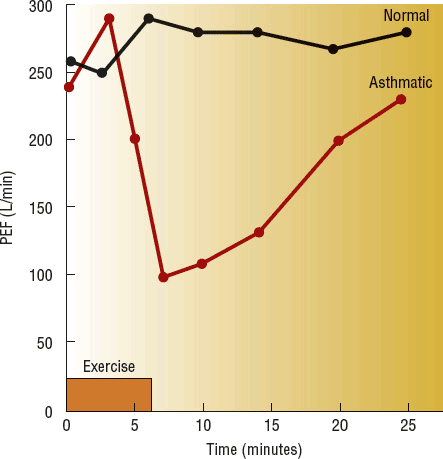
FIGURE 15-3 Typical responses to exercise in a normal subject and an asthmatic subject. Note the initial bronchodilation. (PEF, peak expiratory flow.)
A refractory period following EIB lasts up to 3 hours after exercise in some patients. During this period, repeat exercise of the same intensity produces either no decrease in pulmonary function or a drop of less than 50% of the initial response.13 This refractory period is thought to be caused by an acute depletion of mast cell mediators and time required for their repletion. Patients with known refractoriness to exercise will still respond to histamine, so acute hyporesponsiveness of airway smooth muscle does not appear to be a factor.13
EIB is believed to be a reflection of increased BHR associated with asthma. A correlation, though not perfect, exists between EIB and reactivity to histamine, methacholine, and mannitol.13,17 Other patient groups with BHR (e.g., after viral infection, cystic fibrosis, or allergic rhinitis) show bronchoconstriction after exercise to a lesser degree (5% to 10% drops) than patients with asthma (15% to 40% drops).13 Patients will not always demonstrate the same sensitivity. During periods of remission, a decreased sensitivity to the same degree of exercise is often observed. Finally, a number of children and adults with EIB are otherwise normal, without symptoms or abnormal pulmonary function except in association with exercise.2 Elite athletes have a higher prevalence of EIB than the general population.13
Nocturnal Asthma
![]() Worsening of asthma during sleep is referred to as nocturnal asthma. Patients with nocturnal asthma exhibit significant falls in pulmonary function between bedtime and awakening.2 Typically, their lung function reaches a nadir at 3 to 4 AM. Although the pathogenesis of this phenomenon is unknown, it has been associated with diurnal patterns of endogenous cortisol secretion and circulating epinephrine.2 Direct evidence for an inflammatory component to nocturnal asthma includes increased circulating histamine and activated eosinophils and LT excretion at night associated with increased BHR to methacholine.2
Worsening of asthma during sleep is referred to as nocturnal asthma. Patients with nocturnal asthma exhibit significant falls in pulmonary function between bedtime and awakening.2 Typically, their lung function reaches a nadir at 3 to 4 AM. Although the pathogenesis of this phenomenon is unknown, it has been associated with diurnal patterns of endogenous cortisol secretion and circulating epinephrine.2 Direct evidence for an inflammatory component to nocturnal asthma includes increased circulating histamine and activated eosinophils and LT excretion at night associated with increased BHR to methacholine.2
Numerous other factors that may affect nocturnal worsening of asthma, including allergies and improper environmental control, gastroesophageal reflux, obstructive sleep apnea, and sinusitis, also must be considered when evaluating these patients.2 Most experts consider nocturnal symptoms to be a sign of inadequately treated persistent asthma.2 Awakening from nocturnal asthma is a sensitive indicator of both severity and inadequate control.2
FACTORS CONTRIBUTING TO ASTHMA SEVERITY
Viral Respiratory Infections
Viral respiratory infections are primarily responsible for exacerbations of asthma, particularly in children under age 10.9,10 Infants are particularly susceptible to airway obstruction and wheezing with viral infections because of their small airways. The most common cause of exacerbations in both children and adults is the rhinovirus, which is the most frequent virus associated with the common cold and distributed worldwide.10 Other viruses isolated include RSV, parainfluenza virus, adenoviruses, coronavirus, and influenza viruses. Certain viruses (RSV and parainfluenza virus) are capable of inducing specific IgE antibodies, and rhinovirus can activate eosinophils directly in asthmatics.10 The increase in asthma symptoms and BHR that occurs may last for days or weeks following resolution of the symptoms of the viral infection. Evidence does not support a beneficial effect of influenza vaccine for preventing asthma exacerbations from subsequent influenza infections.2
Environmental and Occupational Factors
Agents and events and the mechanisms that are known to trigger asthma are listed in Table 15-1.2 The general mechanisms are unknown but presumably are the result of epithelial damage and inflammation in the airway mucosa. Ozone and sulfur dioxide, common components of air pollution, have been used to induce BHR in animals. Exposure to 0.2 ppm ozone for 2 to 3 hours can induce bronchoconstriction and increase BHR in asthmatics.2,8 Sulfur dioxide in the ambient atmosphere is highly irritating and presumably induces bronchoconstriction through mast cell or irritant-receptor involvement.2 Asthma produced by repeated prolonged exposure to industrial inhalants is a significant health problem. It has been estimated that occupational asthma accounts for 2% of all asthmatic persons.8 Persons with occupational asthma have the typical symptoms of asthma with cough, dyspnea, and wheeze. Typically, the symptoms are related to workplace exposure and improve on days off and during vacations.8 In some instances, symptoms may persist even after termination of exposure.8
Stress, Depression, and Psychosocial Factors in Asthma
Observational studies demonstrate an association between increased stress and worsening asthma, but the role is not clearly defined.2 Bronchoconstriction from psychological factors appears to be mediated primarily through excess parasympathetic input. Atropine has been shown to block experimental psychogenic bronchoconstriction. It is most important to emphasize to both patients and parents that asthma is not an emotional disease; however, coping skills may benefit the patient who becomes emotionally distraught during an asthma attack.
Rhinitis/Sinusitis
Disorders of the upper respiratory tract, particularly rhinitis and sinusitis, have been linked with asthma for many years. As many as 40% to 50% of asthmatics have abnormal sinus radiographs.2 However, chronic sinusitis may just represent a nonbacterial coexisting condition with allergic asthmatics because the histologic changes in the paranasal sinuses are similar to those seen in the lung and nose.2 Treatment of upper airway disease may optimize overall asthma control. The mechanism by which sinusitis aggravates asthma is unknown. The treatment of allergic rhinitis with intranasal corticosteroids and cromolyn but not antihistamines will reduce BHR in asthmatic patients.2 It has been postulated that transport of mucus chemotactic factors and inflammatory mediators from nasal passages during allergic rhinitis into the lung may accentuate BHR.
Gastroesophageal Reflux Disease
Symptoms of gastroesophageal reflux disease (GERD) as well as asymptomatic reflux are common in both children and adults who have asthma.2 Nocturnal asthma may be associated with nighttime reflux.2 Reflux of acidic gastric contents into the esophagus is thought to initiate a vagally mediated reflex bronchoconstriction.2 Also of concern is that most medications that decrease airway smooth muscle tone may have a relaxant effect on gastroesophageal sphincter tone. However, treatment of reflux in asthma patients has produced inconsistent results on asthma control.2,18 The current recommendation is to initiate standard antireflux therapy in those patients exhibiting symptoms of reflux.2
Female Hormones and Asthma
Premenstrual worsening of asthma has been reported in as many as 30% to 40% of women in some studies, whereas worsening of pulmonary functions has been reported even in women not aware of worsening symptoms.19 The pathophysiology is uncertain because estrogen replacement in postmenopausal women has been shown to worsen asthma, whereas estradiol and progesterone administration has been variably reported to improve or have no effect on asthma in women with premenstrual asthma.19,20 Asthma symptoms may vary significantly during different stages of the menstrual cycle. The clinical significance of menstruation-related asthma is still unclear because some studies have reported that up to 50% of ED visits by women were premenstrual, whereas others have reported no association with menstrual phase.19,20 In general, BHR and symptoms improve in asthmatic women during pregnancy.2,19
FOODS, DRUGS, AND ADDITIVES
Documentation in the literature of food allergens as triggers for asthma is not available.2 However, additives, specifically sulfites used as preservatives, can trigger life-threatening asthma exacerbations. Beer, wine, dried fruit, and open salad bars, in particular, have high concentrations of metabisulfites.2 Severe oral corticosteroid-dependent patients should be warned about ingesting foods processed with sulfites. Another additive producing bronchospasm is benzalkonium chloride, which is found as a preservative in some nebulizer solutions of antiasthmatic drugs.21
Aspirin and other nonsteroidal antiinflammatory drugs can precipitate an attack in up to 20% of adults and 5% of children with asthma.22 The mechanism is related to cyclooxygenase inhibition, and 5-lipoxygenase inhibition can alter dose–response but not completely block the symptoms.22 The prevalence increases with age and severity of asthma.2 The greatest frequency occurs in severe corticosteroid-resistant asthmatics in their fourth and fifth decades who also have perennial rhinitis and nasal polyposis (presence of several polyps).22 Other drugs that do not precipitate bronchospasm but that prevent its reversal are the nonselective β-blocking agents.2
Nutritional Factors
Epidemiologic data suggest that obesity increases the prevalence of asthma and may reduce asthma control.23 Lung volume and tidal volume are reduced in obesity, promoting airway narrowing. Obesity also produces low-grade systemic inflammation that may act on the lung to worsen asthma.23 The mechanism is likely the release of adipose-derived proinflammatory mediators such as IL-6, IL-10, eotaxin, tumor necrosis factor-α, transforming growth factors-β1, C-reactive protein, leptin, and adiponectin or a result of common predisposing dietary factors. Although not all studies find relationship between body mass index and asthma control, management of asthma in obese patients should include weight loss measures.24
More recently it has been shown that children with vitamin D insufficiency are at greater risk of uncontrolled asthma (increased hospitalizations, BHR, and eosinophil counts).25 Vitamin D helps regulate T cells and improves their secretion of antiinflammatory cytokines in response to corticosteroids.25 Clinical trials defining the potential therapeutic role of vitamin D supplementation in asthma management are underway.
TREATMENT
Asthma
Aerosol Therapy for Asthma
![]()
![]() Aerosol delivery of drugs for asthma has the advantage of being site specific and thus enhancing the therapeutic ratio.2,26 Inhalation of short-acting β2-agonists provides more rapid bronchodilation than either parenteral or oral administration, as well as the greatest degree of protection against EIB and other challenges.2 Inhaled corticosteroids (ICSs) have been developed with rapid oral and systemic clearance to enhance lung activity and reduce systemic activity. Specific agents (e.g., cromolyn, formoterol, salmeterol, and ipratropium bromide) are only effective by inhalation.2 Therefore, an understanding of aerosol drug delivery is essential to optimal asthma therapy. Table 15-4 lists the factors determining lung deposition of therapeutic aerosols.
Aerosol delivery of drugs for asthma has the advantage of being site specific and thus enhancing the therapeutic ratio.2,26 Inhalation of short-acting β2-agonists provides more rapid bronchodilation than either parenteral or oral administration, as well as the greatest degree of protection against EIB and other challenges.2 Inhaled corticosteroids (ICSs) have been developed with rapid oral and systemic clearance to enhance lung activity and reduce systemic activity. Specific agents (e.g., cromolyn, formoterol, salmeterol, and ipratropium bromide) are only effective by inhalation.2 Therefore, an understanding of aerosol drug delivery is essential to optimal asthma therapy. Table 15-4 lists the factors determining lung deposition of therapeutic aerosols.
TABLE 15-4 Factors Determining Lung Deposition of Aerosols
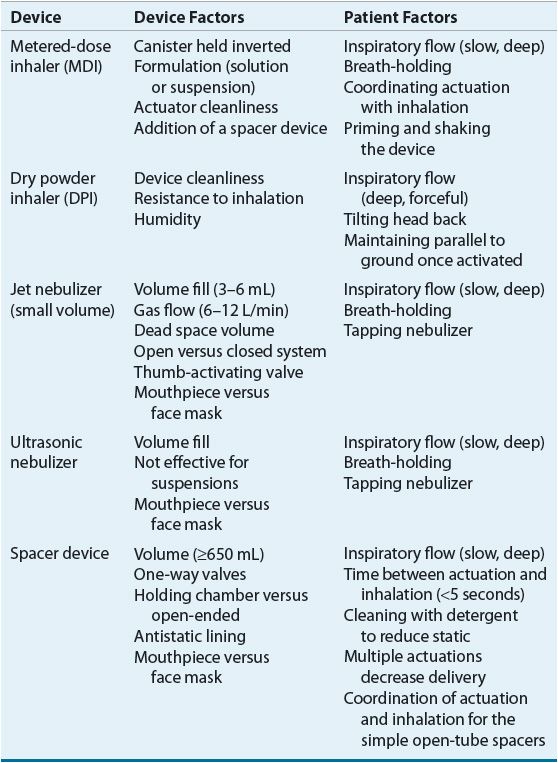
Device Determinants of Delivery
Devices used to generate therapeutic aerosols include jet nebulizers, ultrasonic nebulizers, metered-dose inhalers (MDIs), and dry powder inhalers (DPIs). The single most important device factor determining the site of aerosol deposition is particle size.26 Devices for delivering therapeutic aerosols generate particles with aerodynamic diameters from 0.5 to 35 μm.26 Particles larger than 10 μm deposit in the oropharynx, particles between 5 and 10 μm deposit in the trachea and large bronchi, particles 1 to 5 μm in size reach the lower airways, and particles smaller than 0.5 μm act as a gas and are exhaled. In asthma, the airways, not the alveoli, are the target for delivery. Respirable particles are deposited in the airways by three mechanisms: (a) inertial impaction, (b) gravitational sedimentation, and (c) Brownian diffusion.26 The first two mechanisms are the most important for therapeutic aerosols and probably are the only factors that can be manipulated by patients.
Each delivery device within a classification generates specific aerosol characteristics, so extrapolation of delivery data from one device cannot be applied to the other devices in the class. For instance, MDIs can deliver 15% to 50% of the actuated dose; DPIs, 10% to 30% of the labeled dose; and nebulizers, 2% to 15% of the starting dose.26 MDIs and DPIs are portable and convenient, unlike jet nebulizers. Small portable ultrasonic nebulizers have also been developed. MDIs consist of a pressurized canister with a metering valve; the canister contains active drug, low-vapor-pressure propellants such as hydrofluoroalkane (HFA), cosolvents, and/or surfactants.26 The international ban on the production and use of chlorofluorocarbon (CFC) propellant (due to their ozone layer–depleting properties) has resulted in no CFC-propelled MDIs available in the United States with the exception of pirbuterol Autohaler until December 31, 2013. With any change in the components of an MDI, the FDA considers it to be a new drug that requires stability, safety, and efficacy studies prior to approval. The drug is either in solution or a suspended micronized powder. In order to disperse the suspension for accurate delivery, the canister must be shaken. The metering chamber measures a liquid volume, and, therefore, the device must be held with the valve stem downward so that the chamber is covered with liquid26 (Fig. 15-4). When the canister is actuated, the device releases the propellant and drug in a forceful spray whose particles are large (mass median aerodynamic diameter [MMAD] = 45 μm)26 (see Fig. 15-4). As evaporation occurs, the particle size is reduced to a final MMAD of 0.5 to 5.5 μm depending on the MDI. The aerosol cloud extends about 6 in beyond the MDI at the lowest MMAD.26 Each MDI has different conditions for storage and durations to expiration, so the pharmacist must become familiar with and counsel the patient on these factors.
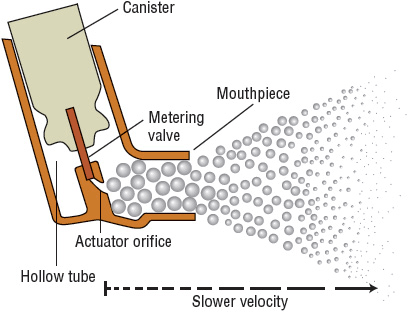
FIGURE 15-4 Illustration of a metered-dose inhaler demonstrating the particle size difference as the aerosol cloud extends outward.
The breath-actuated MDI Autohaler is cocked with a lever to “load” the dose of medication, a baffle is opened by inspiratory pressure, and the dose is expelled from the canister metering chamber.26 While the need for hand–lung coordination for proper actuation is reduced significantly with breath-actuated MDIs, these devices do not accommodate the use of a spacer device.
Spacer devices are used frequently with an MDI to decrease oropharyngeal deposition and enhance lung delivery.2 However, not all spacer devices produce similar effects. The design of spacers varies from simple open-ended tubes that separate the MDI from the mouth to valved holding chambers (VHCs) with one-way valves that open during inhalation (the preferred system). A VHC allows evaporation of the propellant prior to inhalation permitting a greater number of drug particles to achieve a respirable droplet size. VHC use also allows inhalation after actuation of the MDI, obviating the need for good hand–lung coordination.26 Additionally, the large particles that normally would deposit in the oropharynx “rain out” in the spacer.26 All the available spacers significantly reduce oropharyngeal deposition from MDIs, with the VHCs being superior to the open-ended tubes.26 This reduction in oropharyngeal deposition is an important factor in reducing local adverse effects (e.g., hoarseness and thrush) from ICSs.26 The change in lung delivery depends on both the MDI and the drug, where one spacer device may enhance delivery with one MDI preparation and decrease delivery with others.26 The use of VHCs is less likely to enhance delivery from HFA-propelled MDIs. Finally, over time, holding chambers can build up static electricity that attracts small particles to the sides of the chamber, significantly reducing aerosol availability. Some spacers should be washed weekly with household detergent with a single rinse and allowed to drip dry.2 Other VHCs have been developed with antistatic materials.
Dry micronized powders can be inhaled directly into the lung. A number of DPIs are now available for use in the United States.26 Currently, there are no generic DPIs as each drug plus device has its own patent. Each DPI has unique characteristics with advantages and disadvantages (Table 15-5). The primary advantage of DPIs is that they are breath actuated and require minimal hand–lung coordination, and it is thus easier to teach patients proper technique.26 Some DPIs are more flow dependent than others.26 Thus, similar to MDIs and spacers, delivery data from one DPI cannot be extrapolated to another.
TABLE 15-5 Characteristics of Various Inhalation Devices