The major sites of antiviral drug action. Note: interferon-alfas are speculated to have multiple sites of action on viral replication.
(Reproduced, with permission, from Katzung BG, editor: Basic & Clinical Pharmacology, 11th ed. McGraw-Hill, 2009: Fig. 49-1.)
One of the most important trends in viral chemotherapy, especially in the management of HIV infection, has been the introduction of combination drug therapy. This can result in greater clinical effectiveness in viral infections and can also prevent, or delay, the emergence of resistance.
Antiherpes Drugs
Most drugs active against herpes viruses are antimetabolites bioactivated via viral or host cell kinases to form compounds that inhibit viral DNA polymerases.
Acyclovir (Acycloguanosine)
Mechanisms
Acyclovir is a guanosine analog active against herpes simplex virus (HSV-1, HSV-2) and varicella-zoster virus (VZV). The drug is activated to form acyclovir triphosphate, which interferes with viral synthesis in 2 ways. It acts as a competitive substrate for DNA polymerase, and it leads to chain termination after its incorporation into viral DNA (Figure 49-2). Resistance of HSV can involve changes in viral DNA polymerase. However, many resistant strains of HSV (TK- strains) lack thymidine kinase, the enzyme involved in the initial viral-specific phosphorylation of acyclovir. Such strains are cross-resistant to famciclovir, ganciclovir, and valacyclovir.
FIGURE 49-2
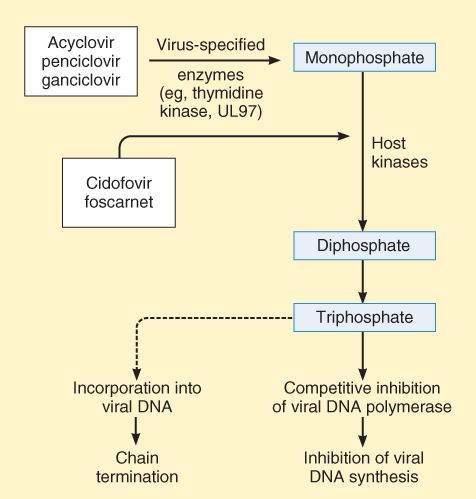
Mechanism of action of antiherpes agents.
(Reproduced, with permission, from Katzung BG, editor: Basic & Clinical Pharmacology, 11th ed. McGraw-Hill, 2009: Fig. 49-3.)
Pharmacokinetics
Acyclovir can be administered by the topical, oral, and intravenous routes. Because of its short half-life, oral administration requires multiple daily doses of acyclovir. Renal excretion is the major route of elimination of acyclovir, and dosage should be reduced in patients with renal impairment.
Clinical Uses and Toxicity
Oral acyclovir is commonly used for the treatment of mucocutaneous and genital herpes lesions (Table 49-1) and for prophylaxis in AIDS and in other immunocompromised patients (eg, those undergoing organ transplantation). The oral drug is well tolerated but may cause gastrointestinal (GI) distress and headache. Intravenous administration is used for severe herpes disease, including encephalitis, and for neonatal HSV infection. Toxic effects with parenteral administration include delirium, tremor, seizures, hypotension, and nephrotoxicity. Acyclovir has no significant toxicity on the bone marrow.
TABLE 49-1 Important antiviral drugs.
Virus Primary Drugs Alternative or Adjunctive Drugs CMV Ganciclovir, valganciclovir Cidofovir, foscarnet, fomiversin HSV, VZV Acyclovira
Cidofovir, foscarnet, vidarabine HBV IFN- , lamivudine Adefovir dipivoxil, entacavir, lamivudine, telbivudine HCV IFN-
, lamivudine Adefovir dipivoxil, entacavir, lamivudine, telbivudine HCV IFN- Ribavirin Influenza A Oseltamivir Amantadine, rimantadine, zanamivir Influenza B Oseltamivir Zanamivir
Ribavirin Influenza A Oseltamivir Amantadine, rimantadine, zanamivir Influenza B Oseltamivir Zanamivir
aAnti-HSV drugs similar to acyclovir include famciclovir, penciclovir, and valacyclovir; IFN- , interferon-
, interferon- .
.
Other Drugs for HSV and VSV Infections
Several newer agents have characteristics similar to acyclovir. Valacyclovir is a prodrug converted to acyclovir by hepatic metabolism after oral administration and reaches plasma leveles 3-5 times greater than those achieved by acyclovir. Valcyclovir has a longer duration of action than acyclovir. Penciclovir undergoes activation by viral thymidine kinase, and the triphosphate form inhibits DNA polymerase but does not cause chain termination. Famciclovir is a prodrug converted to penciclovir by first-pass metabolism in the liver. Used orally in genital herpes and for herpes zoster, famciclovir is well tolerated and is similar to acyclovir in its pharmacokinetic properties. None of the acyclovir congeners has activity against TK- strains of HSV. Docosanol is an aliphatic alcohol that inhibits fusion between the HSV envelope and plasma membranes. It prevents viral entry and subsequent replication. Used topically docosanol shortens healing time.
Ganciclovir
Mechanisms
Ganciclovir, a guanine derivative, is triphosphorylated to form a nucleotide that inhibits DNA polymerases of cytomegalovirus (CMV), and HSV and causes chain termination. The first phosphorylation step is catalyzed by virus-specific enzymes in both CMV-infected and HSV-infected cells. CMV resistance mechanisms involve mutations in the genes that code for the activating viral phosphotransferase and the viral DNA polymerase. Thymidine kinase-deficient HSV strains are resistant to ganciclovir.
Pharmacokinetics
Ganciclovir is usually given intravenously and penetrates well into tissues, including the eye and the central nervous system (CNS). The drug undergoes renal elimination in direct proportion to creatinine clearance. Oral bioavailability is less than 10%. An intraocular implant form of ganciclovir can be used in CMV retinitis. Valganciclovir, a prodrug of ganciclovir, has high oral bioavailability and has decreased the use of intravenous forms of ganciclovir (and also of intravenous cidofovir and foscarnet) in end-organ CMV disease.
Clinical Uses and Toxicity
Ganciclovir is used for the prophylaxis and treatment of CMV retinitis and other CMV infections in immunocompromised patients. Systemic toxic effects include leukopenia, thrombocytopenia, mucositis, hepatic dysfunction, and seizures. The drug may cause severe neutropenia when used with zidovudine or other myelosuppressive agents.
Cidofovir
Mechanisms and Pharmacokinetics
Cidofovir is activated exclusively by host cell kinases and the active diphosphate, which inhibits DNA polymerases of HSV, CMV, adenovirus, and papillomavirus (HPV). Because phosphorylation does not require viral kinase, cidofovir is active against many acyclovir and ganciclovir-resistant strains. Resistance is due to mutations in the DNA polymerase gene. The drug is given intravenously and undergoes renal elimination. Dosage should be adjusted in proportion to creatinine clearance and full hydration maintained.
Clinical Uses and Toxicity
Cidofovir is effective in CMV retinitis, in mucocutaneous HSV infections, including those resistant to acyclovir, and in genital warts. Nephrotoxicity is the major dose-limiting toxicity of cidofovir, additive with other nephrotoxic drugs including amphotericin B and aminoglycoside antibiotics.
Foscarnet
Mechanisms
Foscarnet is a phosphonoformate derivative that does not require phosphorylation for antiviral activity. Although it is not an antimetabolite, foscarnet inhibits viral RNA polymerase, DNA polymerase, and HIV reverse transcriptase. Resistance involves point mutations in the DNA polymerase gene.
Pharmacokinetics
Foscarnet is given intravenously and penetrates well into tissues, including the CNS. The drug undergoes renal elimination in direct proportion to creatinine clearance.
Clinical Uses and Toxicity
The drug is an alternative for prophylaxis and treatment of CMV infections, including CMV retinitis, and has activity against ganciclovir-resistant strains of this virus. Foscarnet inhibits herpes DNA polymerase in acyclovir-resistant strains that are thymidine kinase-deficient and may suppress such resistant herpetic infections in patients with AIDS. Adverse effects are severe and include nephrotoxicity (30% incidence) with disturbances in electrolyte balance (especially hypocalcemia), genitourinary ulceration, and CNS effects (headache, hallucinations, seizures).
Other Antiherpes Drugs
Vidarabine
Vidarabine is an adenine analog and has activity against HSV, VZV, and CMV. Its use for systemic infections is limited by rapid metabolic inactivation and marked toxic potential. Vidarabine is used topically for herpes keratitis but has no effect on genital lesions. Toxic effects with systemic use include GI irritation, paresthesias, tremor, convulsions, and hepatic dysfunction. Vidarabine is teratogenic in animals.
Idoxuridine and Trifluridine
These pyrimidine analogs are used topically in herpes keratitis (HSV-1). They are too toxic for systemic use.
Fomivirsen
Fomivirsen is an antisense oligonucleotide that binds to mRNA of CMV, inhibiting early protein synthesis. The drug is injected intravitreally for treatment of CMV retinitis.
Cross-resistance between fomivirsen and other anti-CMV agents has not been observed. Concurrent systemic anti-CMV therapy is recommended to protect against extraocular and contralateral retinal CMV disease. Fomiversin causes iritis, vitreitis, increased intraocular pressure and changes in vision.
Anti-HIV Drugs
The primary drugs effective against HIV are antimetabolite inhibitors of viral reverse transcriptase and inhibitors of viral aspartate protease (Table 49-2). The current approach to treatment of infection with HIV is the initiation of treatment with 3 or more antiretroviral drugs, if possible, before symptoms appear. Such combinations usually include nucleoside reverse transcriptase inhibitors (NRTIs) together with inhibitors of HIV protease (PI). Highly active antiretroviral therapy (HAART) involving drug combinations can slow or reverse the increases in viral RNA load that normally accompany progression of disease. In many AIDS patients, HAART slows or reverses the decline in CD4 cells and decreases the incidence of opportunistic infections.
TABLE 49-2 Major antiretroviral drugs.
Subclass Prototype Other Significant Agents Nucleoside reverse inhibitors Zidovudine Abacavir, didanosine, emtricitabine, lamivudine, stavudine, zalcitabine, zidovudine Nonnucleoside reverse transcriptase inhibitors Delavirdine Efavirenz, etravirine, nevirapine, tenofovir Protease inhibitors Indinavir Amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, tipranavir CCR-5 antagonist Maraviroc Fusion inhibitor Enfuvirtide
Drug management of HIV infection is subject to change. Updated recommendations can be obtained at the following websites: ATIS, http://www.hivatis.org; and NPIN, http://www.cdcnpin.org.
Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
To convert their RNA into dsDNA, retroviruses require virally encoded RNA-dependent DNA polymerase (reverse transcriptase). Mammalian RNA and DNA polymerases are sufficiently distinct to permit a selective inhibition of the viral reverse transcriptase.
NRTIs are prodrugs converted by host cell kinases to triphosphates, which not only competitively inhibit binding of natural nucleotides to the dNTP-binding site of reverse transcriptase but also act as chain terminators via their insertion into the growing DNA chain. Because NRTIs lack a 3′-hydroxyl group on the ribose ring, attachment of the next nucleotide is impossible. Resistance emerges rapidly when NRTIs are used as single agents via mutations in the pol gene; cross-resistance occurs but is not complete.
Abacavir
A guanosine analog, abacavir has good oral bioavailability and an intracellular half-life of 12-24 h. HIV resistance requires several concomitant mutations and tends to develop slowly. Hypersensitivity reactions, occasionally fatal, occur in 5% of HIV patients.
Didanosine (ddI)
Oral bioavailability of ddI is reduced by food and by chelating agents. The drug is eliminated by the kidney, and the dose must be reduced in patients with renal dysfunction. Pancreatitis is dose-limiting and occurs more frequently in alcoholic patients and those with hypertriglyceridemia. Other adverse effects include peripheral neuropathy, diarrhea, hepatic dysfunction, hyperuricemia and CNS effects.
Emtricitabine
Good oral bioavailability and renal elimination with long half-life permits once-daily dosing of emtricitabine. Because of the propylene glycol in the oral solution, the drug is contraindicated in pregnancy and young children and in patients with hepatic or renal dysfunction. Common adverse effects of the drug include asthenia, GI distress, headache, and hyperpigmentation of the palms and/or the soles.
Lamivudine (3TC)
Lamivudine is 80% bioavailable by the oral route and is eliminated almost exclusively by the kidney. In addition to its use in HAART regimens for HIV, lamivudine is also effective in hepatitis B infections. Dosage adjustment is needed in patients with renal insufficiency. Adverse effects of lamivudine are usually mild and include GI distress, headache, insomnia, and fatigue.
Stavudine (d4T)
Stavudine has good oral bioavailability and penetrates most tissues, including the CNS. Dosage adjustment is needed in renal insufficiency. Peripheral neuropathy is dose-limiting and increased with coadministration of didanosine or zalcitabine. Lactic acidosis with hepatic steatosis occurs more frequently with stavudine than with other NRTIs.
Tenofovir
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


