26.1 INTRODUCTION TO ANTI-VIRAL DRUGS
In this chapter we discuss another major application of virology: the development and use of drugs to combat virus diseases in humans. Drugs are available for the treatment of diseases caused by a variety of viruses, and in some circumstances drugs are also used for the prevention of virus infections. We shall discuss the modes of action of several anti-viral drugs, and their clinical uses. We shall also consider some of the problems associated with anti-viral drug use, especially the emergence of drug-resistant virus strains.
For a long time there were very few anti-viral drugs available for clinical use compared with the number of anti-bacterial and anti-fungal drugs. This was because it was difficult to find compounds that interfere specifically with viral activities without causing significant harm to host cell activities. As we have seen in earlier chapters, however, there are many virus-specific activities that are potential drug targets, some of which are summarized in Figure 26.1.
Figure 26.1 Virus activities that are potential drug targets. A generalized view of virus replication is depicted; no single virus performs all of these activities! DNA replication provides a target if the virus produces enzymes (e.g. DNA polymerase, thymidine kinase) distinct from those of the host.
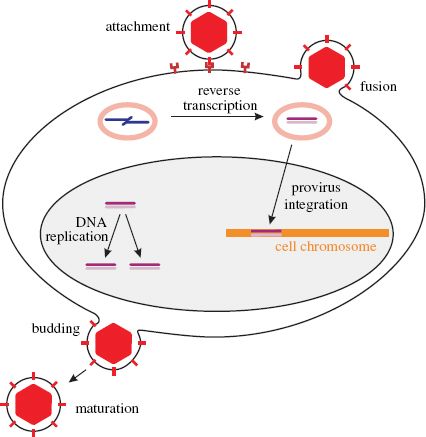
Most of the early anti-viral drugs were found by screening large numbers of compounds, but this approach has now been largely superseded by designing drugs to inhibit specific virus proteins. Both approaches will be outlined in this chapter.
26.2 DEVELOPMENT OF ANTI-VIRAL DRUGS
26.2.1 Screening compounds for anti-viral activity
In this approach to the search for clinically useful anti-viral drugs, large numbers of compounds are tested for possible anti-viral activity. Some of the compounds chosen in the past had already been shown to have anti-tumor cell activity.
The screening procedure involves testing dilutions of the compounds against a range of viruses growing in cell cultures. Evidence that a compound interferes with the replication of a virus might include inhibition of cytopathic effect (CPE) or inhibition of plaque formation. For each potentially useful compound the concentration that inhibits CPE or plaque formation by 50% is determined. This concentration of the drug is known as the 50% inhibitory concentration (IC50).
Screening compounds for anti-viral activity has been largely replaced by the rational design of drugs.
26.2.2 Rational design of anti-viral drugs
The process of designing an anti-viral drug starts with deciding on a virus activity that the drug will target (Figure 26.1). Once this has been selected, it is necessary to choose a target protein, such as a viral enzyme, that is involved in that activity. A detailed picture of the three-dimensional structure of the protein is derived using techniques such as X-ray crystallography (Section 2.5.3), and a target site in the protein is selected. Computer programs are then used to design compounds that will bind to the target site with the aim of inhibiting the activity of the virus protein. Rational design has been used to develop a large number of anti-viral drugs, though the mode of action of some of them remains unclear.
26.2.3 Safety of anti-viral drugs
If a compound is to have clinical value it must not only inhibit virus replication, but it must cause little or no harm to the host. The research and development procedures therefore include incubating uninfected cell cultures with the compound and examining them for damage such as death or inhibition of cell division.
The potential value of a compound can be assessed by determining the ratio of its cell toxicity to its anti-viral activity. This ratio is known as the selectivity index (SI) and is expressed by the formula

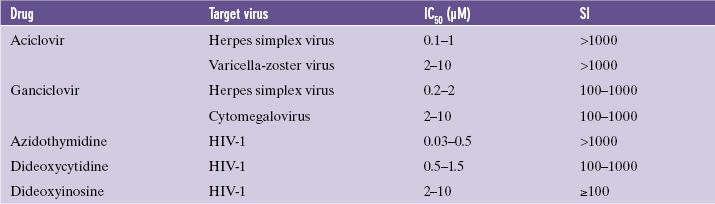
A compound with a low IC50 and a high SI is most likely to have value as an anti-viral drug. Some examples of IC50 and SI values are given in Table 26.1. The data indicate that aciclovir would be preferred to ganciclovir for treatment of herpes simplex virus infections.
Table 26.1 Some anti-viral drugs and target viruses. IC50: 50% inhibitory concentration. SI: selectivity index. IC50 and SI vary depending on the virus strain and the cell type. These figures are ranges determined using several cell types. Data from Kinchington et al. (1995)
26.3 EXAMPLES OF ANTI-VIRAL DRUGS
26.3.1 Nucleoside analogs
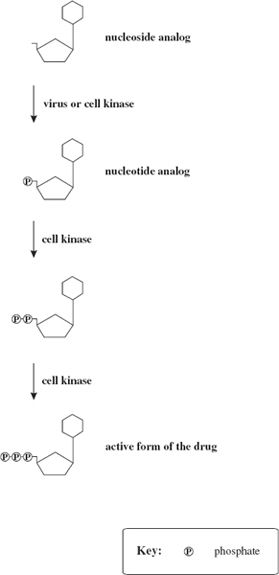
A number of anti-viral drugs are synthetic compounds structurally similar to nucleosides such as guanosine and 2′-deoxythymidine (Figure 26.2) and they act by interfering with the synthesis of virus nucleic acids. After being taken into a cell a nucleoside analog, like a nucleoside, is phosphorylated at the 5′ carbon (or its equivalent) to become a nucleotide analog (Figure 26.3). The 5′ triphosphate derivative of the nucleoside analog is the active form of the drug and acts as a competitive inhibitor of a viral polymerase such as a reverse transcriptase.
Figure 26.2 Nucleoside analogs used as anti-viral drugs. The analogs are derived from the nucleosides shown on the left.
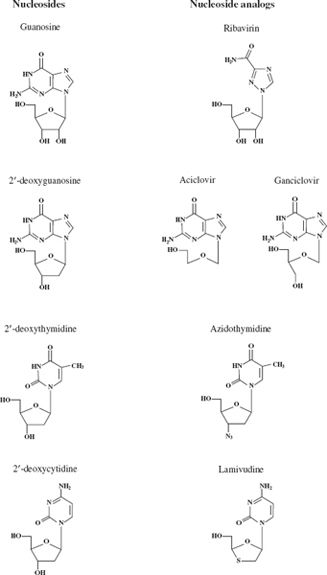
Figure 26.3 Phosphorylation of a nucleoside analog. The active form of the drug is the 5′ triphosphate derivative of the nucleoside analog.
During nucleic acid synthesis, if one of these nucleotide analogs is incorporated into a growing strand, then nucleic acid synthesis is terminated, as in the dideoxy method of DNA sequencing. The structure of the nucleotide analog prevents it from accepting the next nucleotide. In some cases the analog lacks the hydroxyl group on the 3′ carbon (or lacks the 3′ carbon) necessary for the addition of the next nucleotide.
26.3.1.a Ribavirin
Ribavirin is an analog of guanosine (Figure 26.2). It is used for the treatment of infection with several RNA viruses, especially hepatitis C virus (HCV; Section 23.11). Combined treatment with ribavirin and pegylated α-interferon eradicates HCV infection in about 50% of patients, but the treatment often causes significant side effects. Inhibitors of an HCV protease have shown promising results in clinical trials and are to be used, in conjunction with ribavirin and interferon, for treatment of HCV infections.
Ribavirin has been used for treatment of infections with respiratory syncytial virus (Section 15.5), one of the most important causes of severe respiratory tract disease in children. There are, however, doubts about the effectiveness of the drug, and the mode of delivery is expensive and inconvenient (aerosol for 12–18 hours daily), so its use is restricted to some high-risk patients.
The mode of action of ribavirin is not completely understood and several hypotheses have been proposed to explain its anti-viral activity. The drug inhibits the cell enzyme inosine monophosphate dehydrogenase, and has been shown to inhibit several virus activities, including viral RNA synthesis; it is likely that it has more than one mode of action.
26.3.1.b Nucleoside analogs that inhibit herpesvirus DNA synthesis
Aciclovir (also spelt “acyclovir”) is an analog of guanosine (Figure 26.2). It is a very safe drug, with almost no side effects, widely used for the treatment and prevention of diseases caused by three of the herpesviruses: herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) and varicella-zoster virus (VZV) (Chapter 11). Uses of aciclovir include:
- prevention of cold sores;
- prevention and treatment of genital herpes;
- treatment of HSV encephalitis (early treatment reduces the mortality rate);
- protection of immunocompromised patients after exposure to VZV;
- treatment of shingles (pain is reduced significantly).
Aciclovir strongly inhibits virus DNA synthesis but has very little effect on cell DNA synthesis. In a herpesvirus-infected cell the first phosphorylation of aciclovir (Figure 26.3) is carried out by the virus thymidine kinase (TK) (Section 11.5.3). The cell TK is produced only at certain stages of the cell cycle and has a much lower affinity for aciclovir than the viral enzyme, so there is very little phosphorylation of aciclovir in uninfected cells.
During DNA synthesis, aciclovir triphosphate competes with deoxyguanosine triphosphate (dGTP) for incorporation into the new strand. When aciclovir becomes incorporated, DNA synthesis is terminated due to the lack of a 3′—OH group.
Ganciclovir, like aciclovir, is an analog of guanosine, and the first phosphorylation in a cell is carried out by a virus enzyme, in this case a protein kinase. Ganciclovir is more likely than aciclovir to become incorporated into cell DNA. As a result ganciclovir has a lower SI than aciclovir (Table 26.1) and can cause significant side effects, especially reduced numbers of blood cells because of damage to bone marrow cells.
Ganciclovir is used to treat infections with cytomegalovirus, as aciclovir is less effective against this virus than it is against the other herpesviruses. Examples of cytomegalovirus diseases that are treated with ganciclovir include pneumonia and retinitis in immunocompromised patients, such as those who have received organ transplants and those with AIDS.
26.3.1.c Nucleoside analogs that inhibit reverse transcription
Azidothymidine (AZT), also known as zidovudine, is an analog of thymidine (Figure 26.2). Like other nucleoside analogs, it is phosphorylated to the 5′ triphosphate after uptake into a cell (Figure 26.3). AZT had been investigated as a possible anti-cancer drug and later it was found to inhibit the reverse transcriptase of HIV.
AZT triphosphate binds more strongly to the viral reverse transcriptase than to the cell DNA polymerase, and the reverse transcriptase binds AZT triphosphate in preference to deoxythymidine triphosphate. AZT thus inhibits HIV reverse transcription, but it can also interfere with cell DNA synthesis and, like ganciclovir, it can damage the bone marrow. In the early days of AZT treatment HIV-infected patients were administered large doses and many patients suffered severe side effects.
Lamivudine, also known as 3TC, is an analog of cytidine (Figure 26.2). It inhibits reverse transcriptases, including those of HIV and hepatitis B virus (HBV), and is used, usually in combination with other therapies, to treat infections with these viruses. The drug does not eliminate the infections, but it can control them. For treatment of HBV infection lamivudine is often given in combination with α-interferon; after treatment for a year or more inflammation of the liver is reduced in a proportion of patients.
Several other nucleoside analogs that inhibit reverse transcription are used for treatment of HIV infection (examples are dideoxycytidine and dideoxyinosine) or HBV infection (an example is adefovir).
26.3.2 Non-nucleoside inhibitors of reverse transcription
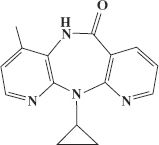
The approach of rational design was used to develop several HIV reverse transcriptase inhibitors that are not based on nucleosides. These non-nucleoside inhibitors target different sites in the reverse transcriptase to those targeted by the nucleoside analogs. An example of a non-nucleoside inhibitor that is used against HIV is nevirapine (Figure 26.4).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


