Angioimmunoblastic T-Cell Lymphoma
Definition
Angioimmunoblastic T-cell lymphoma (AILT) is a specific type of peripheral T-cell lymphoma that is characterized by lymphadenopathy, systemic disease, and associated immunodysregulation and immunodeficiency (1).
Synonyms
Angioimmunoblastic lymphadenopathy with dysproteinemia (AILD); AILD-like lymphoma; AILD-type T-cell lymphoma (Kiel); immunoblastic lymphadenopathy; lymphogranulomatosis X; diffuse mixed small- and large-cell, diffuse large-cell, or large-cell immunoblastic (Working Formulation).
Historical Perspective
This disease was originally described in the German literature by Forster and Moeschlin in 1954 (2). In 1974, these lesions were described in more detail and designated as angioimmunoblastic lymphadenopathy with dysproteinemia (AILD), immunoblastic lymphadenopathy, and lymphogranulomatosis X by others (2,3,4). The term AILD caught on and became generally used. Angioimmunoblastic lymphadenopathy with dysproteinemia was originally thought to be a premalignant lesion, but affected patients had a poor prognosis. Patients were shown to have a high risk of dying from infectious complications, and a subset of patients went on to develop histologically overt lymphoma.
With the advent of routine immunophenotypic, cytogenetic, and molecular techniques being applied to the study of lymphomas, it became clear that a high frequency of cases of AILD had an aberrant immunophenotype, cytogenetic abnormalities, or molecular evidence of T-cell monoclonality. In the 1994 Revised European-American Lymphoma (REAL) Classification it was suggested that AILD is actually malignant at its onset, and therefore was better designated as angioimmunoblastic T-cell lymphoma (5). This was adopted by the World Health Organization (WHO) classification.
Does AILD as originally described still exist? For all practical purposes, AILD no longer exists as an entity. These lesions are now classified as angioimmunoblastic T-cell lymphoma (AILT; also abbreviated as AITL in many studies). However, clonality cannot be demonstrated in approximately 10% of cases, and some patients reported in the literature had a relatively indolent clinical course. Thus, some pathologists are of the opinion that this small subset of cases without evidence of monoclonality, currently accepted as part of AILT, may be a preneoplastic or dysplastic disease, as AILD was originally described (6).
Epidemiology
According to the Nebraska Non-Hodgkin’s Lymphoma Classification Project, AILT represents 1.2% of all non-Hodgkin lymphomas and 18% of all T-cell lymphomas (7). These relative frequencies are best applied to western Europe and North America. In the Surveillance, Epidemiology, and End Results (SEER) data from the United States, from 1992–2001, AILT represented 2.8% of all T/NK-cell lymphomas and 0.15% of all lymphomas (including Hodgkin and unclassified lymphomas) (8). The incidence rate was 0.05 per 100,000 person years. In many (but not all) large clinicopathologic studies there is a slight male predominance (9). Angioimmunoblastic T-cell lymphoma appears to be much more common in whites than in African or Asian Americans (8).
Pathogenesis
Angioimmunoblastic T-cell lymphoma is a monoclonal T-cell neoplasm thought to arise in follicular T-helper cells (10). Normal follicular T-helper cells reside in lymphoid follicles and are essential for the formation and maintenance of follicles. These cells typically express CD4, CD10, BCL-6, and CXCL13, as is true for the neoplastic cells in most cases of AILT (10,11).
Currently, the causes or molecular events involved in the neoplastic transformation of follicular T-helper cells are not understood. Dunleavy et al. (12) have speculated that AILT is an antigen-driven process, with the most likely antigen being Epstein-Barr virus (EBV). This suggestion is based on the common presence of EBV in AILT, often early in the clinical evolution of the disease, and the location of the virus in nonneoplastic B cells associated with these neoplasms. In cases of AILT with hyperplastic lymphoid follicles, the EBV can selectively infect germinal center B cells. Epstein-Barr virus-infected B cells have been shown to present EBV proteins to normal T cells in association with major histocompatibility complex class II antigens thereby upregulating CD28 (13). For these reasons, it seems possible that EBV may be involved in activating follicular T-helper cells (or their neoplastic equivalents), resulting in secretion of CXCL13 and other substances. However, little data currently is available to prove that EBV activates or is involved in neoplastic transformation of follicular T-helper cells in AILT. In addition, we have observed patients who have lymphomas with most or all of the features of AILT, except that the lymphomas are EBV-, who subsequently relapse with AILT associated with EBV+ B-cells. Cases such as these have led to an alternative and more accepted view that EBV+ B-cells are present in AILT as a secondary event, related to immunodeficiency.
It is thought that the neoplastic cells of AILT retain, at least in part or in a chaotic manner, the functional capabilities of follicular T-helper cells. As a result, the neoplastic cells of AILT can secrete a variety of chemokines and cytokines. These substances can induce changes themselves and can also recruit nonneoplastic cells and induce them to secrete chemokines, cytokines, and other molecules. Possibly, an immune regulation loop results
from the effects of neoplastic cells on reactive cells and vice versa. The net effect of these results would be a chemokine and cytokine storm resulting in systemic disease–associated immunodysregulation and immunodeficiency. This would account for many of the symptoms and laboratory abnormalities and the distinctive histologic appearance characterized by a polymorphous cell population and vascular proliferation.
from the effects of neoplastic cells on reactive cells and vice versa. The net effect of these results would be a chemokine and cytokine storm resulting in systemic disease–associated immunodysregulation and immunodeficiency. This would account for many of the symptoms and laboratory abnormalities and the distinctive histologic appearance characterized by a polymorphous cell population and vascular proliferation.
Some the substances secreted by either neoplastic or nonneoplastic cells that could account for the systemic manifestations of AILT include CXCL13, interleukin (IL)-1 and IL-6, tumor necrosis factor, lymphotoxin α, and vascular endothelial growth factor (VEGF)-A (14,15,16). CXCL13 is particularly notable in this group because it is important for the entry of B-cells into lymphoid follicles, it stimulates the expansion of follicular dendritic cells, and it is involved in B-cell development and activation (17). Secretion of VEGF may explain the prominent vascularity in cases of AILT (16).
Immunodeficiency associated with AILT, which can be severe, most likely accounts for the predisposition of these patients to infectious complications and second neoplasms. Since the initial recognition of this disease, when it was known as lymphogranulomatosis X or AILD, patients with AILT have been known to be highly susceptible to infectious complications, and infections (bacterial or fungal) are a major cause of death in these patients (2,3,4). Second neoplasms also occur in patients with AILT, with the most common being diffuse large B-cell lymphoma. The topic of second lymphomas in patients with AILT is discussed separately later in this chapter.
Clinical Findings
Angioimmunoblastic T-cell lymphoma is a disease of adults, most often affecting middle-aged or older persons, with a median age around 60 years of age (1,7,9,18). However, the age range is broad, from 17 to 91 years in two recent studies (9,18). The onset is often rapid; symptoms develop within days or weeks. The mean time from onset of symptoms to diagnosis was 3.6 months in one study (9). Constitutional symptoms, including fever, night sweats, and weight loss, occur in over 75% of patients (9). A poor performance status [Eastern Cooperative Oncology Group (ECOG) ≥2] is common, in approximately half of patients (9,18). Pruritus occurs in 40% to 50% of patients (9). Symptoms related to peripheral cytopenias are common, in 30% to 40%, and cough/dyspnea or arthralgia each occur in approximately 20% of patients. A myriad of other symptoms can occur depending on the organ systems involved by disease.
The physical examination reveals lymphadenopathy in over 90% of patients with AILT (1,7,9,18). Lymph node involvement is almost always generalized, and superficial lymph nodes are commonly involved. Splenomegaly occurs in over 50% of patients, whereas hepatomegaly is detected less frequently, in approximately one-quarter. Other physical manifestations include skin lesions; effusions involving the pleural, pericardial, or abdominal cavities; head and neck involvement (particularly the tonsils); and central or peripheral nervous system manifestations. Most patients have Ann Arbor clinical stage III of IV disease and an intermediate or high International Prognostic Index (3 to 5/5) (9,18).
In many studies, skin lesions in a subset of patients appear to be related to drug hypersensitivity, as they arise following drug administration. However, many different drugs have been implicated, and the skin lesions rarely regress after the drug is discontinued (9). A more likely explanation is that the immune dysregulation and deficiency inherent in patients with AILT facilitate skin manifestations.
A number of laboratory manifestations occur in AILT patients (1,9,18). Anemia is common, in approximately half of patients, most often as a result of autoimmune-based erythrocyte destruction, but also probably related to bone marrow involvement. A positive direct Coombs tests is common, in 50% to 60% of patients. Lymphopenia is common, in approximately 50%, and autoimmune mediated leukopenia and thrombocytopenia occur in approximately 30% and 20% of patients, respectively (9,18). In a subset of patients, leukocytes are increased in AILT patients. In 30% to 40% of patients, atypical plasmacytoid lymphocytes and plasma cells (so-called immunocytes) are identified in the blood (19). Lymphoma cells also can be identified in the peripheral blood in a subset of patients (9). Absolute numbers of eosinophils or neutrophils also can be increased.
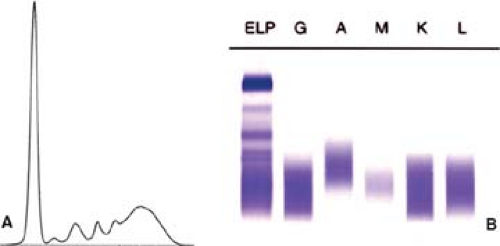 Figure 74.1. Polyclonal hypergammaglobulinemia. A: Tracing of serum proteins. B: Immunofixation showing a polyclonal (smear) pattern with antibodies specific for immunoglobulin heavy and light chains. ELP, electropherogram. (Figure courtesy of Beverly C. Handy, M.D., M.D. Anderson Cancer Center). |
Serum chemistry studies reveal many abnormalities in AILT patients. Most patients have elevated levels of lactate dehydrogenase (LDH) and β-2-microglobulin. Polyclonal hypergammaglobulinemia (Fig. 74.1) is also common, occurring in 51% of patients in one study (9) and 84% of patients in another (18). Monoclonal gammopathy, usually at a low level, also can be present in a small subset of patients (9,20). Acute phase reactants (e.g., C-reactive protein) are commonly elevated. Serum evidence of liver or renal failure is present in approximately 50% and 25% of patients, respectively (9,18). Immune-mediated glomerulonephritis has been reported in patients with AILT (21). Cryoglobulins, cold agglutinins, hypocomplementemia, and autoantibodies (antinuclear, anti–smooth muscle, antithyroperoxidase, rheumatoid factor, etc.) occur in subsets of patients with AILT (9).
The prognosis of patients with AILT is generally poor, and no chemotherapy regimen is currently considered adequate to treat patients with this disease. Many patients respond to chemotherapy, but the complete remission rate is low, approximately 20% (18). Immunomodulating agents have been used on patients in small studies, such as interferon α, rituximab (anti-CD20), thalidomide, bevacizumab (anti-VEGF), and cyclosporine A (22,23,24,25). In a recent study by Lachenal and colleagues (9), the actuarial overall survival at 3 years was 49%, and progression-free survival was 31%. The median survival of AILT patients is 3 years or less (26,27). Nevertheless, occasional patients are reported in the literature who did relatively well treated only with steroids; misdiagnosis of AILT or detection of disease at an early, nonclonal (so-called dysplastic stage) are possible explanations.
Histologic Findings
Lymph Node
The histologic findings in AILT can show remarkable variability in different biopsy specimens (1,2,3,4,7,11,28,29,30,31). However, three features are common to most neoplasms: (a) AILT has a
diffuse pattern and subtotally or completely replaces the lymph node architecture. In cases that partially replace lymph node, AILT has a paracortical distribution. (b) Numerous arborizing blood vessels, usually with thickened walls and lined by hyperplastic endothelial cells, are present in virtually all cases of AILT. Most of these vessels correspond to high endothelial venules. (c) The cell population in AILT is highly polymorphous, with a variable number of neoplastic lymphocytes of small and large size, associated with numerous reactive cells including plasma cells, eosinophils, histiocytes, small lymphocytes, and follicular dendritic cells.
diffuse pattern and subtotally or completely replaces the lymph node architecture. In cases that partially replace lymph node, AILT has a paracortical distribution. (b) Numerous arborizing blood vessels, usually with thickened walls and lined by hyperplastic endothelial cells, are present in virtually all cases of AILT. Most of these vessels correspond to high endothelial venules. (c) The cell population in AILT is highly polymorphous, with a variable number of neoplastic lymphocytes of small and large size, associated with numerous reactive cells including plasma cells, eosinophils, histiocytes, small lymphocytes, and follicular dendritic cells.
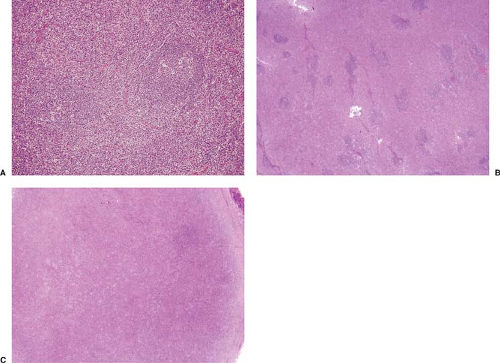 Figure 74.2. Histologic patterns in angioimmunoblastic T-cell lymphoma. A: Pattern I. B: Pattern II. C: Pattern III. A–C, hematoxylin-eosin stain. |
AILT has been subdivided into three histologic patterns, I through III, with more than one pattern commonly existing in the same biopsy specimen. The cut-offs between these patterns are arbitrary (Fig. 74.2 A–C) (28). In pattern I, the overall lymph node architecture is partially preserved and many hyperplastic lymphoid follicles are present. Usually, these follicles have prominent germinal centers but lack well-formed mantle zones (Fig. 2A) (28,31). Relatively normal appearing lymphoid follicles also can be observed. This pattern occurs in 10% to 20% of cases. In cases with this pattern, the infiltrate is highly polymorphous and usually does not overtly resemble lymphoma. In pattern II, the lymph node architecture is mostly effaced but residual, small follicles are present (Fig. 2B). These follicles are often partially disrupted and have irregular shapes. This pattern occurs in up to half of all cases. In pattern III, representing the remainder of cases of AILT, the lymph node architecture is completely replaced and follicles are few or not identified (Fig. 2C). The cell population in patterns II and III tends to be more monotonous and, in some cases, is composed of numerous atypical lymphoma cells, often with clear cytoplasm.
The follicles in patterns II and III show regressive changes, and follicles with advanced regression have been described as “burned out” (Fig. 74.3) (1). These follicles generally lack lymphocytes and show mostly follicular dendritic cells that are disordered. Follicular dendritic cells also proliferate in AILT. This proliferation in AILT can be minor in pattern I but can be very prominent in patterns II and III, both within follicles and
adjacent or surrounding proliferating blood vessels. As these changes are more easily appreciated by immunohistochemical analysis, they are discussed later.
adjacent or surrounding proliferating blood vessels. As these changes are more easily appreciated by immunohistochemical analysis, they are discussed later.
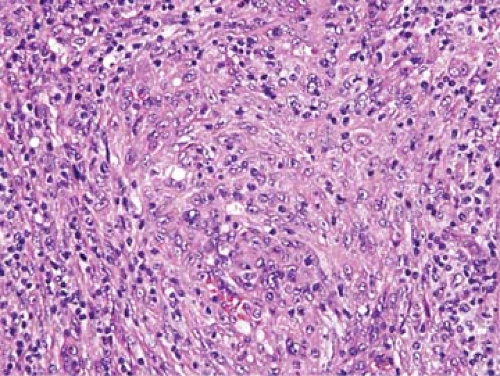 Figure 74.3. Angioimmunoblastic T-cell lymphoma. In this field, a “burned out” germinal center is shown in which numerous follicular dendritic cells and almost no lymphocytes are present. Hematoxylin-eosin stain. |
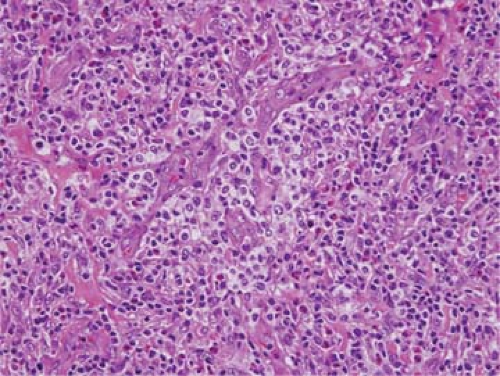 Figure 74.4. Angioimmunoblastic T-cell lymphoma in which neoplastic clear cells surround blood vessels. (A biopsy specimen from the same patient is shown in Fig. 74.14.) Hematoxylin-eosin stain. |
These patterns most likely correspond to stages of disease evolution, with pattern I being early, and pattern III being late. In one recent study in which sequential biopsy specimens were studied in 31 patients with AILT, in seven (23%) patients the lesions progressed from pattern I to pattern III, usually within 2 years (32). In addition, one AILT patient with pattern III disease, after therapy, relapsed with pattern I disease. Other studies with smaller numbers of cases also have observed histologic progression from pattern I to III (29). In general, the number of neoplastic cells increases from pattern I to pattern III. It can be difficult to reliably identify individual neoplastic cells in pattern I cases as the neoplastic cells can represent only a small percentage of the total number of cells in the specimen (analogous to classical Hodgkin lymphoma) (33,34).
Other features have been described in cases of AILT. The neoplastic cells in AILT can have clear cytoplasm, imparting a distinctive appearance; these cells can accumulate around blood vessels or form nodules or tumor masses (Figs. 74.4 and 74.5) (1,30). However, clear cells can be absent in an appreciable subset of AILT cases. In two recent studies, clear cells were identified in only 41% and 66% of neoplasms, respectively (29,35). Clear cells are more commonly identified easily in pattern II or III cases of AILT. Extension through the capsule into perinodal adipose tissue is common in AILT. Nevertheless, it is also common for some of the subcapsular sinuses to remain patent (29,35). These sinuses also can be distended by monocytoid B cells. Clusters of epithelioid histiocytes can be present, and in some cases these cells are numerous, resembling the lymphoepithelioid variant of peripheral T-cell lymphoma, unspecified (PTCL-U; so-called Lennert lymphoma) (36). Plasma cells can be numerous in some cases, and tend to cluster around blood vessels (Fig. 74.6 A–C) (30). Deposits of acidophilic, sludgy material that are positive for the periodic acid–Schiff (PAS) reaction, originally emphasized in early reports of this disease (4), are typically found in approximately 20% to 30% of cases examined at the histologic level (small foci are more commonly detected by electron microscopy) (36). Immunoblasts are present in variable numbers. In occasional AILT cases, large cells with irregularly shaped, polylobated nuclei resembling Reed-Sternberg cells are present; these cells also can resemble Reed-Sternberg cells at the immunophenotypic level (CD15+, CD30+, B-cell antigens+/-) (1,30). Mitotic figures are present in variable numbers in AILT cases, but they can be frequent. Necrosis and fibrosis are usually absent in AILT.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


