Angioimmunoblastic T-cell Lymphoma
Sa A. Wang, MD
Key Facts
Terminology
Peripheral T-cell lymphoma derived from CD4(+) follicular helper T cells characterized by
Lymphadenopathy, systemic disease, and, usually, immunodysregulation and immunodeficiency
Clinical Issues
Advanced stage with generalized lymphadenopathy, hepatomegaly, &/or splenomegaly
Aggressive, with median survival of < 3 years
Anemia, hypereosinophilia, polyclonal hypergammaglobulinemia
Microscopic Pathology
Lymph node
Partial or complete effacement of architecture
Neoplastic T cells with clear/pale cytoplasm
Proliferation of arborizing high endothelial venules
Proliferation of follicular dendritic cells
Ancillary Tests
CD2(+), CD3(+), CD4(+) CD5(+), TCR-αβ(+)
CD10(+/−), Bcl-6(+/−), CXCL13(+/−), PD-1(+/−)
B immunoblasts(+) in variable numbers
Monoclonal T-cell receptor rearrangements
EBER(+) in ˜ 80-90% of cases
Neoplastic cells have gene expression profile of follicular helper T cells
Top Differential Diagnoses
Viral lymphadenitis or drug reaction
Classical Hodgkin lymphoma
T-cell/histiocyte-rich large B-cell lymphoma
Peripheral T-cell lymphoma, not otherwise specified
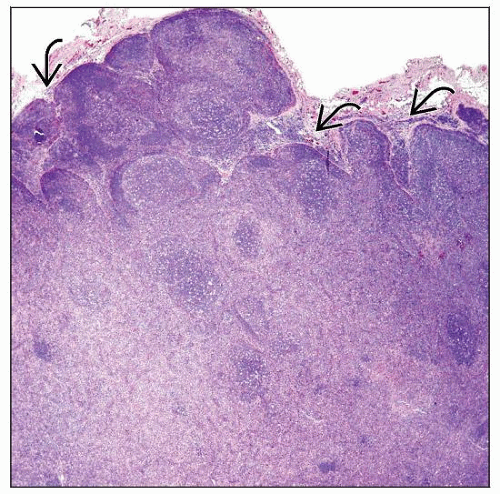 Lymph node involved by angioimmunoblastic T-cell lymphoma (AITL) shows residual follicles, expanded paracortical and interfollicular regions, and open subcapsular (peripheral) sinuses  . . |
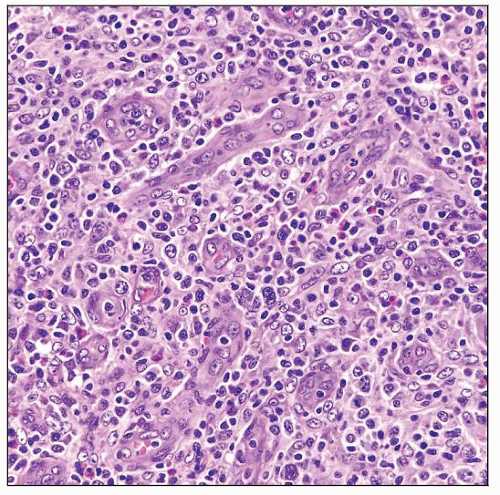 AITL involving lymph node shows increased high endothelial venules (HEV) and a mixed cellular infiltrate. Around the HEV are clusters of neoplastic clear cells. |
TERMINOLOGY
Abbreviations
Angioimmunoblastic T-cell lymphoma (AITL)
Synonyms
Angioimmunoblastic lymphadenopathy with dysproteinemia (AILD)
AILD-like (type) T-cell lymphoma
Immunoblastic lymphadenopathy
Definitions
Peripheral T-cell lymphoma (PTCL) derived from CD4(+) follicular helper T cells characterized by
Lymphadenopathy, systemic disease, and, usually, immunodysregulation and immunodeficiency
ETIOLOGY/PATHOGENESIS
Immunodysregulation
AITL is tumor of follicular helper T cells
Follicular helper T cells upregulate CXCR5 and CXCL13
CXCL13 promotes B-cell recruitment through adherence of B cells on high endothelial venules (HEV)
CD21(+) follicular dendritic cells expand from HEV
Leads to B-cell expansion, plasmacytic differentiation, and hypergammaglobulinemia
Viral Infection
EBV(+) B cells are detected in most cases of AITL
Most likely secondary event as result of host immunocompromise
Human herpesvirus 6B (HHV6B) is also detected by PCR in almost 1/2 of AITL cases
EBV and potentially HHV6B may
Modulate secretion of cytokines and chemokines or expression of membrane receptors
Influence development of tumor microenvironment, favoring disease progression
CLINICAL ISSUES
Epidemiology
Incidence
Rate: 0.5 per 100,000 person years in USA
More common in whites than in African-Americans or Asian-Americans
Represents 1.2% of all non-Hodgkin lymphomas and 18% of all PTCLs
AITL is more common in Europe than in North America or Asia
Age
Median: 59-65 years in various studies
Gender
Likely a slight male predominance (but varies in different studies)
Presentation
Subacute or acute systemic illness
Advanced stage with generalized lymphadenopathy, hepatomegaly, &/or splenomegaly
B symptoms are common (fever, weight loss, night sweats)
Skin rash in > 50% of patients
Generalized or predominantly truncal maculopapular eruption mimicking inflammatory dermatosis
Nodular lesions, plaques, purpura, and urticarial lesions also have been observed
Prior to, concurrent with, or following initial diagnosis of AITL
Other systemic manifestations
Arthralgias or arthritis
Pleural effusions, ascites, &/or edema
Lung, neurologic, or gastrointestinal involvement
Cases of AITL have been reported after administration of antibiotics
Most likely, patients with undetected AITL predisposed to infection requiring antibiotic therapy
Laboratory Tests
Complete blood cell count
Anemia
Cryoglobulins or cold agglutinins
Positive Coombs test in many patients
Hypereosinophilia
Lymphopenia (lymphocytosis is rare)
Thrombocytopenia
Polyclonal hypergammaglobulinemia
Hypoalbuminemia
± autoantibodies
Rheumatoid factor, anti-nuclear factor, anti-smooth muscle
Elevated erythrocyte sedimentation rate
Elevated serum lactate dehydrogenase and β2-microglobulin levels
Treatment
No consensus on optimal therapeutic regimen
For medically eligible patients, combined chemotherapy followed by autologous hematopoietic cell transplantation after achieving remission
For nontransplant candidates, combined chemotherapy
Steroids have role for patients who are not candidates for chemotherapy
Novel, investigational therapies are needed for patients with AITL
Prognosis
Aggressive, with median survival of < 3 years
˜ 30% of patients are long-term survivors
Adverse prognostic factors
Male sex, mediastinal lymphadenopathy, and anemia adversely affect overall survival
Overall immune status also influences survival
Histological features of AITL do not correlate with prognosis
IMAGE FINDINGS
Radiographic Findings
Generalized lymphadenopathy, organ involvement, body effusions
Cannot distinguish AITL from other lymphoma types with disseminated disease
MICROSCOPIC PATHOLOGY
Histologic Features
Lymph node
Partial or complete effacement of architecture; perinodal infiltration common
Paracortical distribution of neoplasm
Peripheral sinuses in lymph node cortex are often patent
Neoplastic cells are small- to medium-sized, with clear to pale cytoplasm, distinct cell membranes, and minimal atypia
Tumor cells often form small clusters around follicles and HEV
Background cells are polymorphous
Variable numbers of small reactive lymphocytes, eosinophils, plasma cells, and histiocytes
± immunoblasts of B-cell lineage; number is variable and can be prominent
± HRS-like cells of B-cell lineage; usually EBV(+)
Marked proliferation of arborizing high endothelial venules (HEV)
Increased proliferation of follicular dendritic cells (FDC), usually surrounding HEV
3 patterns in lymph node have been described
Bone marrow
Nodular or interstitial aggregates in paratrabecular or nonparatrabecular distribution
Neoplastic cells are often small; ± clear cytoplasm; can be difficult to identify
Reactive cells include plasma cells, eosinophils, histiocytes, and B cells
Aggregates are associated with blood vessel proliferation
± EBV(+) cells
Uninvolved bone marrow ± reactive changes associated with AITL
Erythroid hyperplasia, polyclonal plasmacytosis, eosinophilia, fibrosis, or hemophagocytosis
Peripheral blood
Lymphocytosis is uncommon
± atypical lymphocytes or activated lymphocytes (so-called immunocytes)
CD10(+) T cells have been shown in many patients by flow cytometric immunophenotyping
Skin
Changes are variable, may not always result from direct tumor infiltration
Changes can range from nonspecific, mild perivascular dermal lymphocytic infiltrate to, more rarely, overt lymphoma
Body effusions
Nonneoplastic in nature and their cause is poorly understood
Mixture of small lymphocytes, histiocytes, ± eosinophils
Morphological variants of AITL
Epithelioid variant
High content of epithelioid histiocytes in small, poorly defined clusters (Lennert-like reaction)
Clear cell-rich variant
Overt lymphomatous proliferation with sheets of neoplastic cells with clear/pale cytoplasm
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


