247 | Amebiasis and Infection with Free-Living Amebas |
AMEBIASIS
DEFINITION
Amebiasis is an infection with the intestinal protozoan Entamoeba histolytica. About 90% of infections are asymptomatic, and the remaining 10% produce a spectrum of clinical syndromes ranging from dysentery to abscesses of the liver or other organs.
LIFE CYCLE AND TRANSMISSION
E. histolytica is acquired by ingestion of viable cysts from fecally contaminated water, food, or hands. Food-borne exposure is most prevalent and is particularly likely when food handlers are shedding cysts or food is being grown with feces-contaminated soil, fertilizer, or water. Besides the drinking of contaminated water, less common means of transmission include oral and anal sexual practices and—in rare instances—direct rectal inoculation through colonic irrigation devices. Motile trophozoites are released from cysts in the small intestine and, in most patients, remain as harmless commensals in the large bowel. After encystation, infectious cysts are shed in the stool and can survive for several weeks in a moist environment. In some patients, the trophozoites invade either the bowel mucosa, causing symptomatic colitis, or the bloodstream, causing distant abscesses of the liver, lungs, or brain. The trophozoites may not encyst in patients with active dysentery, and motile hematophagous trophozoites are frequently present in fresh stools. Trophozoites are rapidly killed by exposure to air or stomach acid, however, and therefore cannot transmit infection.
EPIDEMIOLOGY
![]() About 10% of the world’s population is infected with Entamoeba, the majority with noninvasive Entamoeba dispar. Amebiasis results from infection with E. histolytica and is the third most common cause of death from parasitic disease (after schistosomiasis and malaria). Invasive colitis and liver abscesses are sevenfold more common among men than among women; this difference has been attributed to a disparity in complement-mediated killing. The wide spectrum of clinical disease caused by Entamoeba is due in part to the differences between these two infecting species. E. histolytica has unique isoenzymes, surface antigens, DNA markers, and virulence properties that distinguish it from other genetically related and morphologically identical species, such as E. dispar and E. moshkovskii.
About 10% of the world’s population is infected with Entamoeba, the majority with noninvasive Entamoeba dispar. Amebiasis results from infection with E. histolytica and is the third most common cause of death from parasitic disease (after schistosomiasis and malaria). Invasive colitis and liver abscesses are sevenfold more common among men than among women; this difference has been attributed to a disparity in complement-mediated killing. The wide spectrum of clinical disease caused by Entamoeba is due in part to the differences between these two infecting species. E. histolytica has unique isoenzymes, surface antigens, DNA markers, and virulence properties that distinguish it from other genetically related and morphologically identical species, such as E. dispar and E. moshkovskii.
Most asymptomatic carriers, including men who have sex with men (MSM) and patients with AIDS, harbor E. dispar and have self-limited infections. In this respect, E. dispar is dissimilar to other enteric pathogens such as Cryptosporidium and Cystoisospora belli, which can cause self-limited illnesses in immunocompetent hosts but devastating diarrhea in patients with AIDS. These observations indicate that E. dispar is incapable of causing invasive disease. Unlike E. dispar, E. histolytica can cause invasive disease, as demonstrated in recent reports from Korea, China, and India that suggest higher prevalences of amebic seroconversion, invasive amebiasis, and amebic liver abscesses among HIV-positive than HIV-negative patients. In another study, 10% of asymptomatic patients who were colonized with E. histolytica went on to develop amebic colitis, while the rest remained asymptomatic and cleared the infection within 1 year.
The potential of E. moshkovskii to cause diarrhea, weight loss, and colitis was recently demonstrated in a mouse model of cecal infection. However, the pathogenic potential of this species is not clear. A prospective evaluation of children from the Mirpur community of Dhaka, Bangladesh, found that most children who had diarrheal diseases associated with E. moshkovskii were simultaneously infected with at least one other enteric pathogen.
Areas of highest incidence of Entamoeba infection (due to inadequate sanitation and crowding) include most developing countries in the tropics, particularly Mexico, India, and nations of Central and South America, tropical Asia, and Africa. In a 4-year follow-up study of preschool children in a highly endemic area of Bangladesh, 80% of children had at least one episode of E. histolytica infection and 53% had more than one episode. Naturally acquired immunity did develop but was usually short-lived and correlated with the presence in the stool of secretory IgA antibody to the major adherence lectin galactose N-acetylgalactosamine (Gal/GalNAc). The main groups at risk for amebiasis in developed countries are returned travelers, recent immigrants, MSM, military personnel, and inmates of institutions. Data from the GeoSentinel Surveillance Network, which come from tropical medicine clinics on six continents, showed that, among long-term travelers (trip duration, >6 months), diarrhea due to E. histolytica was among the most common diagnoses.
PATHOGENESIS AND PATHOLOGY
Both trophozoites (Fig. 247-1) and cysts (Fig. 247-2) are found in the intestinal lumen, but only trophozoites of E. histolytica invade tissue. The trophozoite is 20–60 μm in diameter and contains vacuoles and a nucleus with a characteristic central nucleolus. In animals, depletion of intestinal mucus, diffuse inflammation, and disruption of the epithelial barrier precede trophozoite contact with the colonic mucosa. Trophozoites attach to colonic mucus and epithelial cells by their Gal/GalNAc lectin. The earliest intestinal lesions are microulcerations of the mucosa of the cecum, sigmoid colon, or rectum that release erythrocytes, inflammatory cells, and epithelial cells. Proctoscopy reveals small ulcers with heaped-up margins and normal intervening mucosa (Fig. 247-3A). Submucosal extension of ulcerations under viable-appearing surface mucosa causes the classic “flask-shaped” ulcer containing trophozoites at the margins of dead and viable tissues. Although neutrophilic infiltrates may accompany the early lesions in animals, human intestinal infection is marked by a paucity of inflammatory cells, probably in part because of the killing of neutrophils by trophozoites (Fig. 247-3B). Treated ulcers characteristically heal with little or no scarring. Occasionally, however, full-thickness necrosis and perforation occur.
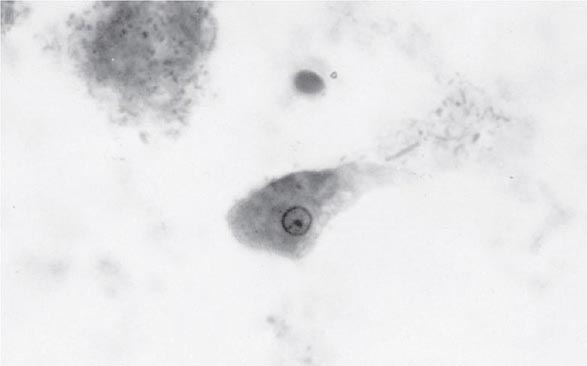
FIGURE 247-1 Trophozoite of E. histolytica. A single nucleus with a central, dot-like nucleolus is seen (trichrome stain).
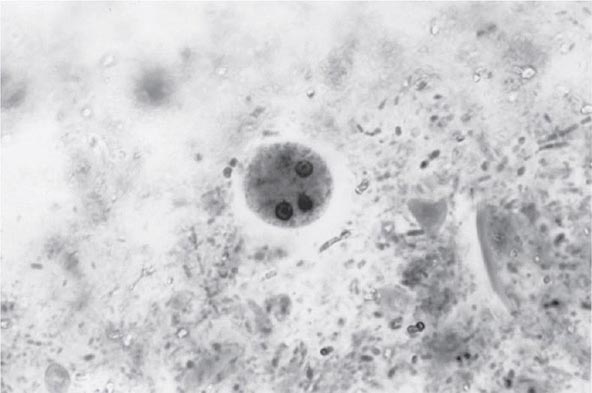
FIGURE 247-2 Cyst of E. histolytica. Three of the four nuclei are visible (trichrome stain).
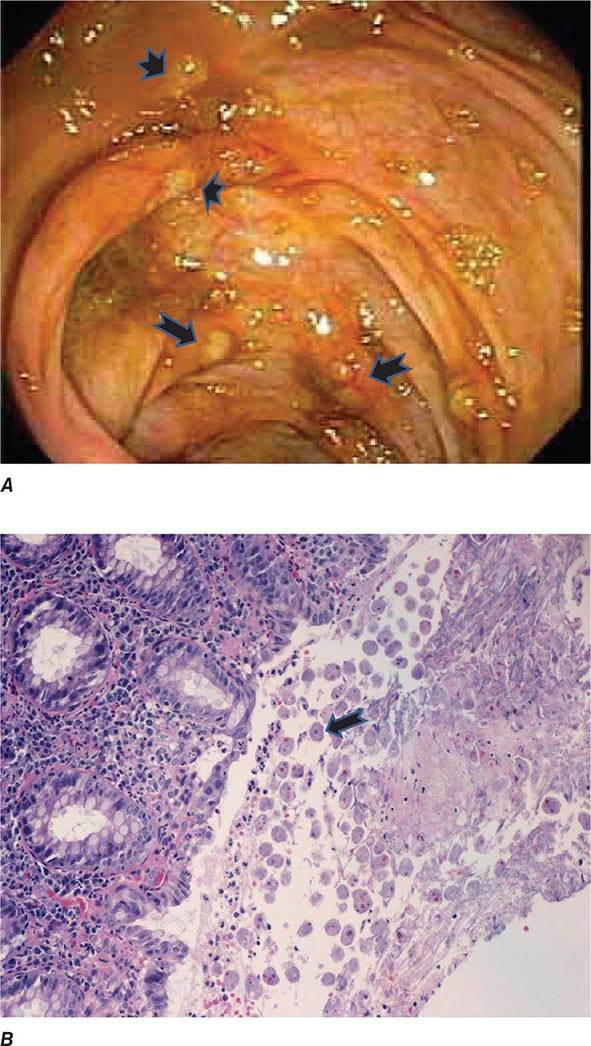
FIGURE 247-3 Endoscopic and histopathologic features of intestinal amebiasis. A. Appearance of ulcers on colonoscopy (arrows). B. Inflammatory infiltrate and E. histolytica trophozoites (arrow) in invasive amebic colitis (hematoxylin and eosin). (Courtesy of the Department of Pathology and Gastroenterology, VA San Diego Medical Center.)
Rarely, intestinal infection results in the formation of a mass lesion, or ameboma, in the bowel lumen. The overlying mucosa is usually thin and ulcerated, while other layers of the wall are thickened, edematous, and hemorrhagic; this condition results in exuberant formation of granulation tissue with little fibrous-tissue response.
A number of virulence factors have been linked to the ability of E. histolytica to invade through the interglandular epithelium. One factor consists of the extracellular cysteine proteinases that degrade collagen, elastin, IgA, IgG, and the anaphylatoxins C3a and C5a. Other enzymes may disrupt glycoprotein bonds between mucosal epithelial cells in the gut. Amebas can lyse neutrophils, monocytes, lymphocytes, and cells of colonic and hepatic lines. The cytolytic effect of amebas appears to require direct contact with target cells and may be linked to the release of phospholipase A and pore-forming peptides. E. histolytica trophozoites also cause apoptosis of human cells. Phagocytosis is a virulence factor that leads to defective parasite proliferation if inhibited. This process is potentially modulated by calmodulin-like calcium-binding protein 3, which pairs with actin and myosin during initiation and formation of phagosomes. Another virulence factor is the ability to resist reactive oxygen species, reactive nitrogen species such as nitric oxide, or S-nitrosothiols such as S-nitrosoglutathione (GSNO) and S-nitrosocysteine (CySNO). E. histolytica trophozoites are constantly exposed to reactive oxygen and nitrogen species from their own metabolism and host defenses during tissue invasion. Overexpression of hydrogen peroxide regulatory motif–binding protein appears to increase E. histolytica cytotoxicity. Since E. histolytica lacks glutathione and glutathione reductase, it relies on its thioredoxin/thioredoxin reductase system to prevent, regulate, and repair the damage caused by oxidative stress. This antioxidant system is versatile in that it can reduce reactive nitrogen species and use an alternative electron donor such as the reduced form of nicotinamide adenine dinucleotide. Metronidazole, the current standard of therapy for amebiasis, seems to exert its antiparasitic effect through the inhibition of this antioxidant system. Newer therapeutic candidates targeting this system, such as auranofin, also have demonstrated in vitro and in vivo efficacy against this parasite.
Liver abscesses are always preceded by intestinal colonization, which may be asymptomatic. Blood vessels may be compromised early by wall lysis and thrombus formation. Trophozoites invade veins to reach the liver through the portal venous system. E. histolytica is resistant to complement-mediated lysis—a property critical to survival in the bloodstream. In contrast, E. dispar is rapidly lysed by complement and is thus restricted to the bowel lumen. Inoculation of amebas into the portal system of hamsters results in an acute cellular infiltrate consisting predominantly of neutrophils. Later, the neutrophils are lysed by contact with amebas, and the release of neutrophil toxins may contribute to necrosis of hepatocytes. The liver parenchyma is replaced by necrotic material that is surrounded by a thin rim of congested liver tissue. The necrotic contents of a liver abscess are classically described as “anchovy paste,” although the fluid is variable in color and is composed of bacteriologically sterile granular debris with few or no cells. Amebas, if seen, tend to be found near the capsule of the abscess.
Host innate and adaptive immunity are important factors that determine susceptibility to invasive disease and its clinical outcome. While neutrophils were thought to contribute to tissue damage in intestinal and liver amebiasis due to their cytotoxic effects on host epithelial cells, a recent report suggests that they may exert a protective effect in susceptible mice. Neutropenia, induced with an antibody to Gr-1 (i.e., to peripheral neutrophils), led to death in C3H/HeJ mice and to severe disease in CBA mice (both of which are relatively susceptible to E. histolytica infection), while it had no effect on C57BL/6 mice, which are known for their intrinsic resistance to infection with this parasite.
Antimicrobial peptides, such as cathelicidins, are an important part of innate immunity and are induced by E. histolytica upon intestinal invasion in a mouse model. In this model, cecal cathelicidin-related antimicrobial peptide (CRAMP) mRNA increased more than fourfold by 3 days and more than 100-fold at 7 days. However, E. histolytica remained resistant to cathelicidin-mediated killing, probably because the antimicrobial peptide was digested by amebic cysteine proteinases.
![]() IgA plays a critical role in acquired immunity to E. histolytica. A study in Bangladeshi schoolchildren revealed that an intestinal IgA response to Gal/GalNAc reduced the risk of new E. histolytica infection by 64%. Serum IgG antibody is not protective; titers correlate with the duration of illness rather than with the severity of disease. Indeed, Bangladeshi children with a serum IgG response were more likely than those without such a response to develop new E. histolytica infection. In infants from this same Bangladeshi community, passive immunity conferred by maternal parasite-specific IgA via breastfeeding resulted in a 39% reduction in risk of infection and a 64% reduction in risk of diarrheal disease from E. histolytica during the first year of life.
IgA plays a critical role in acquired immunity to E. histolytica. A study in Bangladeshi schoolchildren revealed that an intestinal IgA response to Gal/GalNAc reduced the risk of new E. histolytica infection by 64%. Serum IgG antibody is not protective; titers correlate with the duration of illness rather than with the severity of disease. Indeed, Bangladeshi children with a serum IgG response were more likely than those without such a response to develop new E. histolytica infection. In infants from this same Bangladeshi community, passive immunity conferred by maternal parasite-specific IgA via breastfeeding resulted in a 39% reduction in risk of infection and a 64% reduction in risk of diarrheal disease from E. histolytica during the first year of life.
A link between nutrition and immunity is demonstrated by the elevated rate of infections due to protozoan parasites, including E. histolytica, among undernourished children in developing countries. Resistance to amebiasis is associated with a polymorphism in the receptor for the adipocytokine leptin. Children in a Bangladeshi cohort with a mutant R223 leptin receptor allele were nearly four times more likely to be infected with E. histolytica than those carrying the ancestral Q223 allele. This mutant allele is overrepresented in many geographic areas with a high prevalence of amebiasis, such as Bangladesh and India.
CLINICAL SYNDROMES
Intestinal Amebiasis The most common type of amebic infection is asymptomatic cyst passage. Even in highly endemic areas, most patients harbor E. dispar.
Symptomatic amebic colitis develops 2–6 weeks after the ingestion of infectious E. histolytica cysts. A gradual onset of lower abdominal pain and mild diarrhea is followed by malaise, weight loss, and diffuse lower abdominal or back pain. Cecal involvement may mimic acute appendicitis. Patients with full-blown dysentery may pass 10–12 stools per day. The stools contain little fecal material and consist mainly of blood and mucus. In contrast to those with bacterial diarrhea, fewer than 40% of patients with amebic dysentery are febrile. Virtually all patients have heme-positive stools.
More fulminant intestinal infection, with severe abdominal pain, high fever, and profuse diarrhea, is rare and occurs predominantly in children. Patients may develop toxic megacolon, in which there is severe bowel dilation with intramural air. Patients receiving glucocorticoids are at risk for severe amebiasis. Uncommonly, patients develop a chronic form of amebic colitis, which can be confused with inflammatory bowel disease. The association between severe amebiasis complications and glucocorticoid therapy emphasizes the importance of excluding amebiasis when inflammatory bowel disease is suspected. An occasional patient presents with only an asymptomatic or tender abdominal mass caused by an ameboma, which is easily confused with cancer on barium studies. A positive serologic test or biopsy can prevent unnecessary surgery in this setting. The syndrome of post–amebic colitis—i.e., persistent diarrhea following documented cure of amebic colitis—is controversial; no evidence of recurrent amebic infection can be found, and re-treatment usually has no effect.
Amebic Liver Abscess Extraintestinal infection by E. histolytica most often involves the liver. Of travelers who develop an amebic liver abscess after leaving an endemic area, 95% do so within 5 months. Young patients with an amebic liver abscess are more likely than older patients to present in the acute phase with prominent symptoms of <10 days’ duration. Most patients are febrile and have right-upper-quadrant pain, which may be dull or pleuritic in nature and may radiate to the shoulder. Point tenderness over the liver and right-sided pleural effusion are common. Jaundice is rare. Although the initial site of infection is the colon, fewer than one-third of patients with an amebic abscess have active diarrhea. Older patients from endemic areas are more likely to have a subacute course lasting 6 months, with weight loss and hepatomegaly. About one-third of patients with chronic presentations are febrile. Thus, the clinical diagnosis of an amebic liver abscess may be difficult to establish because the symptoms and signs are often nonspecific. Since 10–15% of patients present only with fever, amebic liver abscess must be considered in the differential diagnosis of fever of unknown origin (Chap. 26).
Complications of Amebic Liver Abscess Pleuropulmonary involvement, which is reported in 20–30% of patients, is the most frequent complication of amebic liver abscess. Manifestations include sterile effusions, contiguous spread from the liver, and rupture into the pleural space. Sterile effusions and contiguous spread usually resolve with medical therapy, but frank rupture into the pleural space requires drainage. A hepatobronchial fistula may cause cough productive of large amounts of necrotic material that may contain amebas. This dramatic complication carries a good prognosis. Abscesses that rupture into the peritoneum may present as an indolent leak or an acute abdomen and require both percutaneous catheter drainage and medical therapy. Rupture into the pericardium, usually from abscesses of the left lobe of the liver, carries the gravest prognosis; it can occur during medical therapy and requires surgical drainage.
Other Extraintestinal Sites The genitourinary tract may become involved by direct extension of amebiasis from the colon or by hematogenous spread of the infection. Painful genital ulcers, characterized by a punched-out appearance and profuse discharge, may develop secondary to extension from either the intestine or the liver. Both of these conditions respond well to medical therapy. Cerebral involvement has been reported in fewer than 0.1% of patients in large clinical series. Symptoms and prognosis depend on the size and location of the lesion.
DIAGNOSTIC TESTS
Laboratory Diagnosis Stool examinations, serologic tests, and noninvasive imaging of the liver are the most important procedures in the diagnosis of amebiasis. Fecal findings suggestive of amebic colitis include a positive test for heme, a paucity of neutrophils, and amebic cysts or trophozoites. The definitive diagnosis of amebic colitis is made by the demonstration of hematophagous trophozoites of E. histolytica (Fig. 247-1). Because trophozoites are killed rapidly by water, drying, or barium, it is important to examine at least three fresh stool specimens. Examination of a combination of wet mounts, iodine-stained concentrates, and trichrome-stained preparations of fresh stool and concentrates for cysts (Fig. 247-2) or trophozoites (Fig. 247-1) confirms the diagnosis in 75–95% of cases. Culture of amebas is more sensitive, but this diagnostic method is not routinely available. If stool examinations are negative, sigmoidoscopy with biopsy of the edge of ulcers may increase the yield, but this procedure is dangerous during fulminant colitis because of the risk of perforation. Trophozoites in a biopsy specimen from a colonic mass confirm the diagnosis of ameboma, but trophozoites are rare in liver aspirates because they are found in the abscess capsule and not in the readily aspirated necrotic center. Accurate diagnosis requires experience, since the trophozoites may be confused with neutrophils and the cysts must be differentiated morphologically from those of Entamoeba hartmanni, Entamoeba coli, and Endolimax nana, which do not cause clinical disease and do not warrant therapy. Unfortunately, the cysts of E. histolytica cannot be distinguished microscopically from those of E. dispar or E. moshkovskii. Therefore, the microscopic diagnosis of E. histolytica can be made only by the detection of Entamoeba trophozoites that have ingested erythrocytes. In terms of sensitivity, stool diagnostic tests based on the detection of the Gal/GalNAc lectin of E. histolytica compare favorably with the polymerase chain reaction and with isolation in culture followed by isoenzyme analysis.
Serology is an important addition to the methods used for parasitologic diagnosis of invasive amebiasis. Enzyme-linked immunosorbent assays and agar gel diffusion assays are positive in more than 90% of patients with colitis, amebomas, or liver abscess. Positive results in conjunction with the appropriate clinical syndrome suggest active disease because serologic findings usually revert to negative within 6–12 months. Even in highly endemic areas such as South Africa, fewer than 10% of asymptomatic individuals have a positive amebic serology. The interpretation of the indirect hemagglutination test is more difficult because titers may remain positive for as long as 10 years.
Up to 10% of patients with acute amebic liver abscess may have negative serologic findings; in suspected cases with an initially negative result, testing should be repeated in 1 week. In contrast to carriers of E. dispar, most asymptomatic carriers of E. histolytica develop antibodies. Thus, serologic tests are helpful in assessing the risk of invasive amebiasis in asymptomatic, cyst-passing individuals in nonendemic areas. Serologic tests also should be performed in patients with ulcerative colitis before the institution of glucocorticoid therapy to prevent the development of severe colitis or toxic megacolon owing to unsuspected amebiasis.
Routine hematology and chemistry tests usually are not very helpful in the diagnosis of invasive amebiasis. About three-fourths of patients with an amebic liver abscess have leukocytosis (>10,000 cells/μL); this condition is particularly likely if symptoms are acute or complications have developed. Invasive amebiasis does not elicit eosinophilia. Anemia, if present, is usually multifactorial. Even with large liver abscesses, liver enzyme levels are normal or minimally elevated. The alkaline phosphatase level is most often elevated and may remain so for months. Aminotransferase elevations suggest acute disease or a complication.
Radiographic Studies Radiographic barium studies are potentially dangerous in acute amebic colitis. Amebomas are usually identified first by a barium enema, but biopsy is necessary for differentiation from carcinoma.
Radiographic techniques such as ultrasonography, CT, and MRI are all useful for detection of the round or oval hypoechoic cyst of an amebic liver abscess. More than 80% of patients who have had symptoms for >10 days have a single abscess of the right lobe of the liver (Fig. 247-4). Approximately 50% of patients who have had symptoms for <10 days have multiple abscesses. Findings associated with complications include large abscesses (>10 cm) in the superior part of the right lobe, which may rupture into the pleural space; multiple lesions, which must be differentiated from pyogenic abscesses; and lesions of the left lobe, which may rupture into the pericardium. Because abscesses resolve slowly and may increase in size in patients who are responding clinically to therapy, frequent follow-up ultrasonography may prove confusing. Complete resolution of a liver abscess within 6 months can be anticipated in two-thirds of patients, but 10% may have persistent abnormalities for a year.
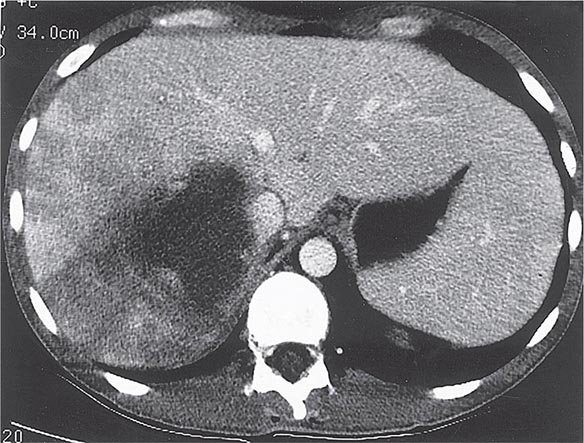
FIGURE 247-4 Abdominal CT scan of a large amebic abscess of the right lobe of the liver. (Courtesy of the Department of Radiology, UCSD Medical Center, San Diego; with permission.)
Differential Diagnosis The differential diagnosis of intestinal amebiasis includes bacterial diarrheas (Chap. 160) caused by Campylobacter (Chap. 192); enteroinvasive Escherichia coli (Chap. 186); and species of Shigella (Chap. 191), Salmonella (Chap. 190), and Vibrio (Chap. 193). Although the typical patient with amebic colitis has less prominent fever than in these other conditions as well as heme-positive stools with few neutrophils, correct diagnosis requires bacterial cultures, microscopic examination of stools, and amebic serologic testing. As has already been mentioned, amebiasis must be ruled out in any patient thought to have inflammatory bowel disease.
Because of the variety of presenting signs and symptoms, amebic liver abscess can easily be confused with pulmonary or gallbladder disease or with any febrile illness with few localizing signs, such as malaria (Chap. 248) or typhoid fever (Chap. 190). The diagnosis should be considered in members of high-risk groups who have recently traveled outside the United States (Chap. 149) and in inmates of institutions. Once radiographic studies have identified an abscess in the liver, the most important differential diagnosis is between amebic and pyogenic abscess. Patients with pyogenic abscess typically are older and have a history of underlying bowel disease or recent surgery. Amebic serology is helpful, but aspiration of the abscess, with Gram’s staining and culture of the material, may be required for differentiation of the two diseases.
TREATMENT | AMEBIASIS |
INTESTINAL DISEASE
The drugs used to treat amebiasis can be classified according to their primary site of action (Table 247-1). Luminal amebicides are poorly absorbed; they reach high concentrations in the bowel, but their activity is limited to cysts and trophozoites close to the mucosa. Only two luminal drugs are available in the United States: iodoquinol and paromomycin. Indications for the use of luminal agents include eradication of cysts in patients with colitis or a liver abscess and treatment of asymptomatic carriers. The majority of asymptomatic individuals who pass cysts are colonized with E. dispar, which does not warrant specific therapy. However, it is prudent to treat asymptomatic individuals who pass cysts unless E. dispar colonization can be definitively demonstrated by specific antigen-detection tests.
DRUG THERAPY FOR AMEBIASIS |
Tissue amebicides reach high concentrations in the blood and tissue after oral or parenteral administration. The development of nitroimidazole compounds, especially metronidazole, was a major advance in the treatment of invasive amebiasis. Patients with amebic colitis should be treated with IV or oral metronidazole. Side effects include nausea, vomiting, abdominal discomfort, and a disulfiram-like reaction. Another longer-acting imidazole compound, tinidazole, is also effective and available in the United States. All patients should also receive a full course of therapy with a luminal agent, since metronidazole does not eradicate cysts. Resistance to metronidazole has been selected in the laboratory but has not been found in clinical isolates. Relapses are not uncommon and probably represent reinfection or failure to eradicate amebas from the bowel because of an inadequate dosage or duration of therapy.
AMEBIC LIVER ABSCESS
Metronidazole is the drug of choice for amebic liver abscess. Longer-acting nitroimidazoles (tinidazole and ornidazole) have been effective as single-dose therapy in developing countries. With early diagnosis and therapy, mortality rates from uncomplicated amebic liver abscess are <1%. There is no evidence that combined therapy with two drugs is more effective than the single-drug regimen. Studies of South Africans with liver abscesses demonstrated that 72% of patients without intestinal symptoms had bowel infection with E. histolytica; thus, all treatment regimens should include a luminal agent to eradicate cysts and prevent further transmission. Amebic liver abscess recurs rarely.
More than 90% of patients respond dramatically to metronidazole therapy with decreases in both pain and fever within 72 h. Indications for aspiration of liver abscesses are (1) the need to rule out a pyogenic abscess, particularly in patients with multiple lesions; (2) the lack of a clinical response in 3–5 days; (3) the threat of imminent rupture; and (4) the need to prevent rupture of left-lobe abscesses into the pericardium. There is no evidence that aspiration, even of large abscesses (up to 10 cm), accelerates healing. Percutaneous drainage may be successful even if the liver abscess has already ruptured. Surgery should be reserved for instances of bowel perforation and rupture into the pericardium.
PREVENTION
Amebic infection is spread by ingestion of food or water contaminated with cysts. Since an asymptomatic carrier may excrete up to 15 million cysts per day, prevention of infection requires adequate sanitation and eradication of cyst carriage. In high-risk areas, infection can be minimized by the avoidance of unpeeled fruits and vegetables and the use of bottled water. Because cysts are resistant to readily attainable levels of chlorine, disinfection by iodination (tetraglycine hydroperiodide) is recommended. There is no effective prophylaxis.
INFECTION WITH FREE-LIVING AMEBAS
EPIDEMIOLOGY
![]() Free-living amebas of the genera Acanthamoeba and Naegleria are distributed throughout the world and have been isolated from a wide variety of fresh and brackish water, including that from lakes, taps, hot springs, swimming pools, and heating and air-conditioning units, and even from the nasal passages of healthy children. Encystation may protect the protozoa from desiccation and food deprivation. The persistence of Legionella pneumophila in water supplies may be attributable in part to chronic infection of free-living amebas, particularly Naegleria. Free-living amebas of the genus Balamuthia have been isolated from soil samples, including a sample from a flowerpot linked to a fatal infection in a child.
Free-living amebas of the genera Acanthamoeba and Naegleria are distributed throughout the world and have been isolated from a wide variety of fresh and brackish water, including that from lakes, taps, hot springs, swimming pools, and heating and air-conditioning units, and even from the nasal passages of healthy children. Encystation may protect the protozoa from desiccation and food deprivation. The persistence of Legionella pneumophila in water supplies may be attributable in part to chronic infection of free-living amebas, particularly Naegleria. Free-living amebas of the genus Balamuthia have been isolated from soil samples, including a sample from a flowerpot linked to a fatal infection in a child.
NAEGLERIA INFECTIONS
Primary amebic meningoencephalitis caused by Naegleria fowleri follows the aspiration of water contaminated with trophozoites or cysts or the inhalation of contaminated dust, leading to invasion of the olfactory neuroepithelium. Infection is most common among otherwise healthy children or young adults, who often report recent swimming in lakes or heated swimming pools. Rarely, some cases occur when contaminated water is used for nasal irrigation. After an incubation period of 2–15 days, severe headache, high fever, nausea, vomiting, and meningismus develop. Photophobia and palsies of the third, fourth, and sixth cranial nerves are common. Rapid progression to seizures and coma may follow. The prognosis is uniformly poor: most patients die within a week. Recently, two surviving children were treated with miltefosine, an investigational drug that is available through the Centers for Disease Control and Prevention (CDC) for the treatment of Naegleria infections.
The diagnosis of Naegleria infection should be considered in any patient who has purulent meningitis without evidence of bacteria on Gram’s staining, antigen detection assay, and culture. Other laboratory findings resemble those for fulminant bacterial meningitis, with elevated intracranial pressure, high white blood cell counts (up to 20,000/μL), and elevated protein concentrations and low glucose levels in cerebrospinal fluid (CSF). Diagnosis depends on the detection of motile trophozoites in wet mounts of fresh spinal fluid. Antibodies to Naegleria species have been detected in healthy adults; serologic testing is not useful in the diagnosis of acute infection.
ACANTHAMOEBA INFECTIONS
Granulomatous Amebic Encephalitis Infection with Acanthamoeba species follows a more indolent course and typically occurs in chronically ill or debilitated patients. Risk factors include lymphoproliferative disorders, chemotherapy, glucocorticoid therapy, lupus erythematosus, and AIDS. Infection usually reaches the central nervous system hematogenously from a primary focus in the sinuses, skin, or lungs. In the central nervous system, the onset is insidious, and the syndrome often mimics a space-occupying lesion. Altered mental status, headache, and stiff neck may be accompanied by focal findings such as cranial nerve palsies, ataxia, and hemiparesis. Cutaneous ulcers or hard nodules containing amebas are frequently detected in AIDS patients with disseminated Acanthamoeba infection and can be an important diagnostic site.
Examination of the CSF for trophozoites may be diagnostically helpful, but lumbar puncture may be contraindicated because of increased intracerebral pressure. CT frequently reveals cortical and subcortical lesions of decreased density consistent with embolic infarcts. In other patients, multiple enhancing lesions with edema may mimic the computed tomographic appearance of toxoplasmosis (Chap. 253). Demonstration of the trophozoites and cysts of Acanthamoeba on wet mounts or in biopsy specimens establishes the diagnosis. Culture on nonnutrient agar plates seeded with Escherichia coli also may be helpful. Fluorescein-labeled antiserum is available from the CDC for the detection of protozoa in biopsy specimens. Granulomatous amebic encephalitis in patients with AIDS may have an accelerated course (with survival for only 3–40 days) because of poor granuloma formation in these individuals. Various antimicrobial agents have been used to treat Acanthamoeba infection, but the infection is almost uniformly fatal. The CDC has now made miltefosine available because of improved survival rates when the drug is included in treatment regimens.
Keratitis The incidence of keratitis caused by Acanthamoeba has increased in the past 30 years, in part as a result of improved diagnosis. Earlier infections were associated with trauma to the eye and exposure to contaminated water. At present, most infections are linked to extended-wear contact lenses, and rare cases are associated with laser-assisted in situ keratomileusis (LASIK). Risk factors include the use of homemade saline, the wearing of lenses while swimming, and inadequate disinfection. Since contact lenses presumably cause microscopic trauma, the early corneal findings may be nonspecific. The first symptoms usually include tearing and the painful sensation of a foreign body. Once infection is established, progression is rapid; the characteristic clinical sign is an annular, paracentral corneal ring representing a corneal abscess. Deeper corneal invasion and loss of vision may follow.
The differential diagnosis includes bacterial, mycobacterial, and herpetic infection. The irregular polygonal cysts of Acanthamoeba (Fig. 247-5) may be identified in corneal scrapings or biopsy material, and trophozoites can be grown on special media. Cysts are resistant to available drugs, and the results of medical therapy have been disappointing. Some reports have suggested partial responses to propamidine isethionate eyedrops. Severe infections usually require keratoplasty.
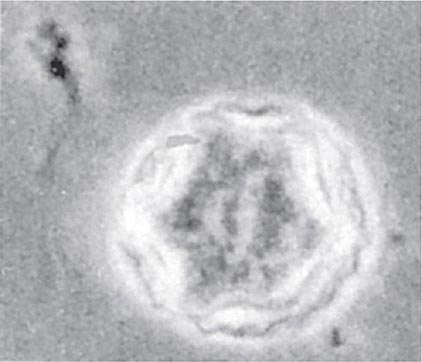
FIGURE 247-5 Double-walled cyst of Acanthamoeba castellanii, as seen by phase-contrast microscopy. (From DJ Krogstad et al, in A Balows et al [eds]: Manual of Clinical Microbiology, 5th ed. Washington, DC, American Society for Microbiology, 1991.)
BALAMUTHIA INFECTIONS
Balamuthia mandrillaris, a free-living ameba previously referred to as a leptomyxid ameba, is an important etiologic agent of amebic meningoencephalitis in immunocompetent hosts. The course is typically subacute, with focal neurologic signs, fever, seizures, and headaches leading to death within 1 week to several months after onset. Examination of CSF reveals mononuclear or neutrophilic pleocytosis, elevated protein levels, and normal to low glucose concentrations. Multiple hypodense lesions are usually detected with imaging studies (Fig. 247-6). This mixed picture of space-occupying lesions with CSF pleocytosis is suggestive of Balamuthia. Fluorescent antibody is available from the CDC for brain biopsy specimens. The variety of drugs used to treat the few surviving patients (i.e., fewer than five reported in the United States) includes pentamidine, flucytosine, sulfadiazine, and macrolides. The CDC recommends that miltefosine now be included, as for the other free-living amebas. The differential diagnosis includes tuberculomas (Chap. 202) and neurocysticercosis (Chap. 260).
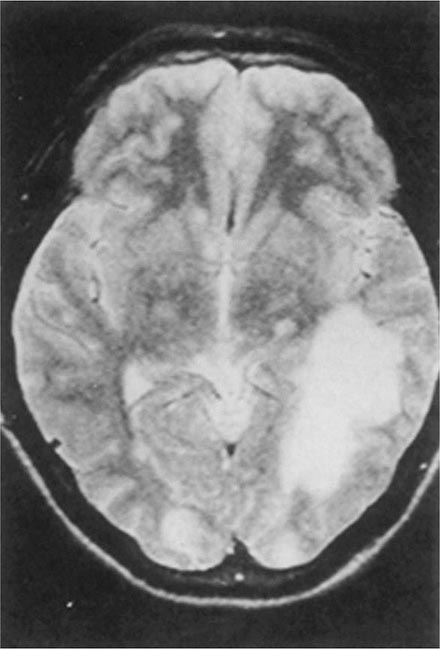
FIGURE 247-6 Brain MRI of amebic meningoencephalitis due to Balamuthia mandrillaris. A large lesion in the parieto-occipital lobe and other smaller lesions are seen. (Courtesy of the Department of Radiology, UCSD Medical Center, San Diego.)
248 | Malaria |
Humanity has but three great enemies: Fever, famine, and war; of these by far the greatest, by far the most terrible, is fever.
—William Osler
Malaria is a protozoan disease transmitted by the bite of infected Anopheles mosquitoes. The most important of the parasitic diseases of humans, it is transmitted in 106 countries containing 3 billion people and causes approximately 2000 deaths each day; mortality rates are decreasing as a result of highly effective control programs in several countries. Malaria has been eliminated from the United States, Canada, Europe, and Russia; in the late twentieth and early twenty-first centuries, however, its prevalence rose in many parts of the tropics. Increases in the drug resistance of the parasite, the insecticide resistance of its vectors, and human travel and migration have contributed to this resurgence. Occasional local transmission after importation of malaria has occurred in several southern and eastern areas of the United States and in Europe, indicating the continual danger to nonmalarious countries. Although there are many successful new control initiatives as well as promising research initiatives, malaria remains today, as it has been for centuries, a heavy burden on tropical communities, a threat to nonendemic countries, and a danger to travelers.
ETIOLOGY AND PATHOGENESIS
Six species of the genus Plasmodium cause nearly all malarial infections in humans. These are P. falciparum, P. vivax, two morphologically identical sympatric species of P. ovale (as suggested by recent evidence), P. malariae, and—in Southeast Asia—the monkey malaria parasite P. knowlesi (Table 248-1). While almost all deaths are caused by falciparum malaria, P. knowlesi and occasionally P. vivax also can cause severe illness. Human infection begins when a female anopheline mosquito inoculates plasmodial sporozoites from its salivary gland during a blood meal (Fig. 248-1). These microscopic motile forms of the malaria parasite are carried rapidly via the bloodstream to the liver, where they invade hepatic parenchymal cells and begin a period of asexual reproduction. By this amplification process (known as intrahepatic or preerythrocytic schizogony or merogony), a single sporozoite eventually may produce from 10,000 to >30,000 daughter merozoites. The swollen infected liver cells eventually burst, discharging motile merozoites into the bloodstream. These merozoites then invade the red blood cells (RBCs) and multiply six- to twentyfold every 48 h (P. knowlesi, 24 h; P. malariae, 72 h). When the parasites reach densities of ~50/μL of blood (~100 million parasites in the blood of an adult), the symptomatic stage of the infection begins. In P. vivax and P. ovale infections, a proportion of the intrahepatic forms do not divide immediately but remain inert for a period ranging from 3 weeks to ≥1 year before reproduction begins. These dormant forms, or hypnozoites, are the cause of the relapses that characterize infection with these two species.
CHARACTERISTICS OF PLASMODIUM SPECIES INFECTING HUMANS |
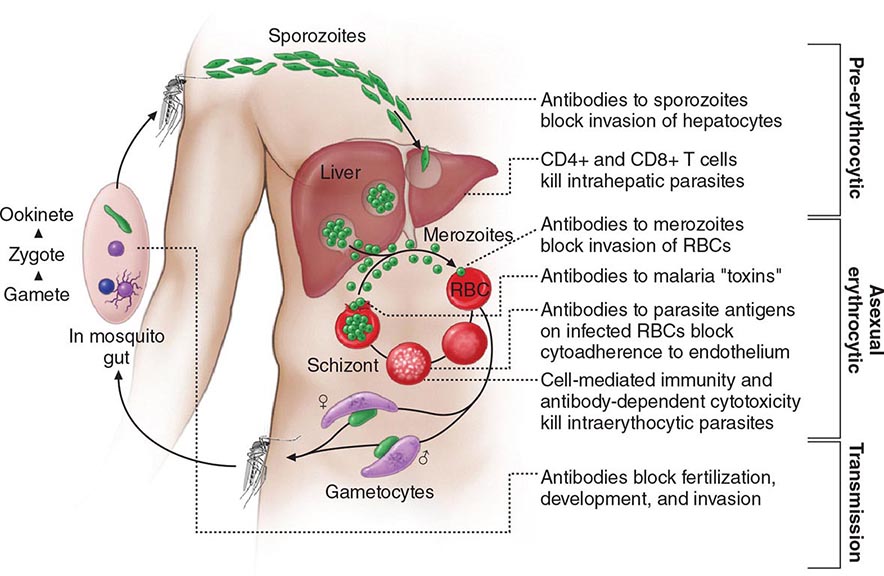
FIGURE 248-1 The malaria transmission cycle from mosquito to human and targets of immunity. RBC, red blood cell.
After entry into the bloodstream, merozoites rapidly invade erythrocytes and become trophozoites. Attachment is mediated via a specific erythrocyte surface receptor. For P. falciparum, the reticulocyte-binding protein homologue 5 (PfRh5) is indispensable for erythrocyte invasion. Basigin (CD147, EMMPRIN) is the erythrocyte receptor of PfRh5. In the case of P. vivax, this receptor is related to the Duffy blood-group antigen Fya or Fyb. Most West Africans and people with origins in that region carry the Duffy-negative FyFy phenotype and are therefore resistant to P. vivax malaria. During the early stage of intraerythrocytic development, the small “ring forms” of the different parasitic species appear similar under light microscopy. As the trophozoites enlarge, species-specific characteristics become evident, pigment becomes visible, and the parasite assumes an irregular or ameboid shape. By the end of the intraerythrocytic life cycle, the parasite has consumed two-thirds of the RBC’s hemoglobin and has grown to occupy most of the cell. It is now called a schizont. Multiple nuclear divisions have taken place (schizogony or merogony). The RBC then ruptures to release 6–30 daughter merozoites, each potentially capable of invading a new RBC and repeating the cycle. The disease in human beings is caused by the direct effects of the asexual parasite—RBC invasion and destruction—and by the host’s reaction. After release from the liver (P. vivax, P. ovale, P. malariae, P. knowlesi), some of the blood-stage parasites develop into morphologically distinct, longer-lived sexual forms (gametocytes) that can transmit malaria. In falciparum malaria, a delay of several asexual cycles precedes this switch to gametocytogenesis.
After being ingested in the blood meal of a biting female anopheline mosquito, the male and female gametocytes form a zygote in the insect’s midgut. This zygote matures into an ookinete, which penetrates and encysts in the mosquito’s gut wall. The resulting oocyst expands by asexual division until it bursts to liberate myriad motile sporozoites, which then migrate in the hemolymph to the salivary gland of the mosquito to await inoculation into another human at the next feeding.
EPIDEMIOLOGY
Malaria occurs throughout most of the tropical regions of the world (Fig. 248-2). P. falciparum predominates in Africa, New Guinea, and Hispaniola (i.e., the Dominican Republic and Haiti); P. vivax is more common in Central America. The prevalence of these two species is approximately equal in South America, the Indian subcontinent, eastern Asia, and Oceania. P. malariae is found in most endemic areas, especially throughout sub-Saharan Africa, but is much less common. P. ovale is relatively unusual outside of Africa and, where it is found, comprises <1% of isolates. Patients infected with P. knowlesi have been identified on the island of Borneo and, to a lesser extent, elsewhere in Southeast Asia, where the main hosts, long-tailed and pig-tailed macaques, are found.
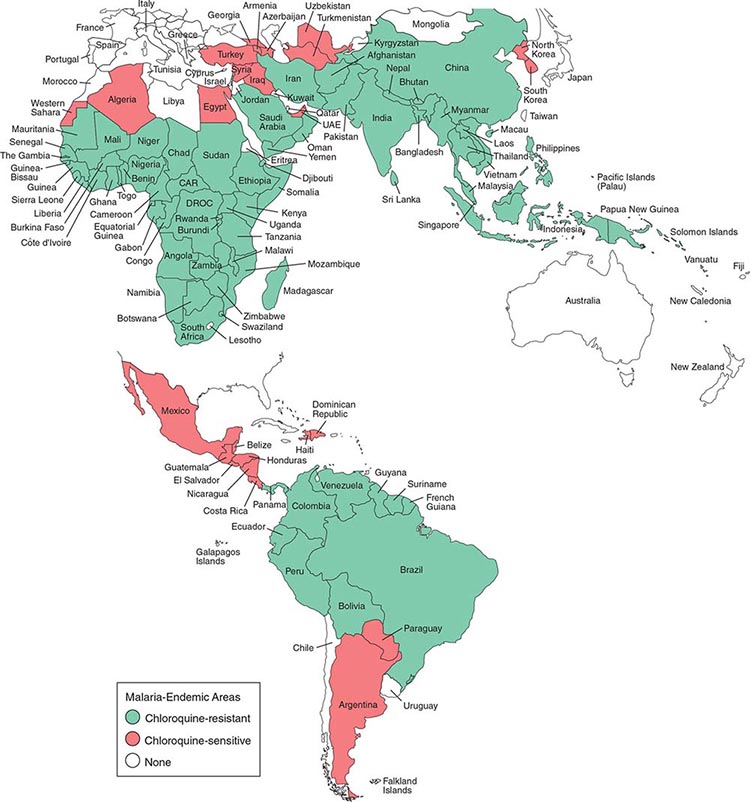
FIGURE 248-2 Malaria-endemic countries in the Americas (bottom) and in Africa, the Middle East, Asia, and the South Pacific (top), 2007. CAR, Central African Republic; DROC, Democratic Republic of the Congo; UAE, United Arab Emirates. Several countries in the Americas, the Middle East, and North Africa are close to eliminating malaria.
The epidemiology of malaria is complex and may vary considerably even within relatively small geographic areas. Endemicity traditionally has been defined in terms of parasitemia rates or palpable-spleen rates in children 2–9 years of age and classified as hypoendemic (<10%), mesoendemic (11–50%), hyperendemic (51–75%), and holoendemic (>75%). Until recently, it was uncommon to use these indices for planning control programs; however, many countries are now conducting national surveys to assess program progress. In holo- and hyperendemic areas (e.g., certain regions of tropical Africa or coastal New Guinea) where there is intense P. falciparum transmission, people may sustain more than one infectious mosquito bite per day and are infected repeatedly throughout their lives. In such settings, rates of morbidity and mortality due to malaria are considerable during early childhood. Immunity against disease is hard won in these areas, and the burden of disease in young children is high; by adulthood, however, most malarial infections are asymptomatic. As control measures progress and urbanization expands, environmental conditions become less conducive to transmission, and all age groups may lose protective immunity and become susceptible to illness. Constant, frequent, year-round infection is termed stable transmission. In areas where transmission is low, erratic, or focal, full protective immunity is not acquired, and symptomatic disease may occur at all ages. This situation usually exists in hypoendemic areas and is termed unstable transmission. Even in stable-transmission areas, there is often an increased incidence of symptomatic malaria coinciding with increased mosquito breeding and transmission during the rainy season. Malaria can behave like an epidemic disease in some areas, particularly those with unstable malaria, such as northern India (the Punjab region), the horn of Africa, Rwanda, Burundi, southern Africa, and Madagascar. An epidemic can develop when there are changes in environmental, economic, or social conditions, such as heavy rains following drought or migrations (usually of refugees or workers) from a nonmalarious region to an area of high transmission, along with failure to invest in national programs; a breakdown in malaria control and prevention services caused by war or civil disorder can intensify epidemic conditions. This situation usually results in considerable mortality among all age groups.
The principal determinants of the epidemiology of malaria are the number (density), the human-biting habits, and the longevity of the anopheline mosquito vectors. More than 100 of the >400 anopheline species can transmit malaria, but the ~40 species that do so commonly vary considerably in their efficiency as malaria vectors. More specifically, the transmission of malaria is directly proportional to the density of the vector, the square of the number of human bites per day per mosquito, and the tenth power of the probability of the mosquito’s surviving for 1 day. Mosquito longevity is particularly important because the portion of the parasite’s life cycle that takes place within the mosquito—from gametocyte ingestion to subsequent inoculation (sporogony)—lasts 8–30 days, depending on ambient temperature; thus, to transmit malaria, the mosquito must survive for >7 days. Sporogony is not completed at cooler temperatures—i.e., <16°C (60.8°F) for P. vivax and <21°C (69.8°F) for P. falciparum; thus transmission does not occur below these temperatures or at high altitudes, although malaria outbreaks and transmission have occurred in the highlands (>1500 m) of eastern Africa, which were previously free of vectors. The most effective mosquito vectors of malaria are those, such as Anopheles gambiae in Africa, that are long-lived, occur in high densities in tropical climates, breed readily, and bite humans in preference to other animals. The entomologic inoculation rate (i.e., the number of sporozoite-positive mosquito bites per person per year) is the most common measure of malaria transmission and varies from <1 in some parts of Latin America and Southeast Asia to >300 in parts of tropical Africa.
ERYTHROCYTE CHANGES IN MALARIA
After invading an erythrocyte, the growing malarial parasite progressively consumes and degrades intracellular proteins, principally hemoglobin. The potentially toxic heme is detoxified by lipid-mediated crystallization to biologically inert hemozoin (malaria pigment). The parasite also alters the RBC membrane by changing its transport properties, exposing cryptic surface antigens, and inserting new parasite-derived proteins. The RBC becomes more irregular in shape, more antigenic, and less deformable.
In P. falciparum infections, membrane protuberances appear on the erythrocyte’s surface 12–15 h after the cell’s invasion. These “knobs” extrude a high-molecular-weight, antigenically variant, strain-specific erythrocyte membrane adhesive protein (PfEMP1) that mediates attachment to receptors on venular and capillary endothelium—an event termed cytoadherence. Several vascular receptors have been identified, of which intercellular adhesion molecule 1 is probably the most important in the brain, chondroitin sulfate B in the placenta, and CD36 in most other organs. Thus, the infected erythrocytes stick inside and eventually block capillaries and venules. At the same stage, these P. falciparum–infected RBCs may also adhere to uninfected RBCs (to form rosettes) and to other parasitized erythrocytes (agglutination). The processes of cytoadherence, rosetting, and agglutination are central to the pathogenesis of falciparum malaria. They result in the sequestration of RBCs containing mature forms of the parasite in vital organs (particularly the brain), where they interfere with microcirculatory flow and metabolism. Sequestered parasites continue to develop out of reach of the principal host defense mechanism: splenic processing and filtration. As a consequence, only the younger ring forms of the asexual parasites are seen circulating in the peripheral blood in falciparum malaria, and the level of peripheral parasitemia underestimates the true number of parasites within the body. Severe malaria is also associated with reduced deformability of the uninfected erythrocytes, which compromises their passage through the partially obstructed capillaries and venules and shortens RBC survival.
In the other human malarias, sequestration does not occur, and all stages of the parasite’s development are evident on peripheral-blood smears. Whereas P. vivax, P. ovale, and P. malariae show a marked predilection for either young RBCs (P. vivax, P. ovale) or old cells (P. malariae) and produce a level of parasitemia that is seldom >2%, P. falciparum can invade erythrocytes of all ages and may be associated with very high levels of parasitemia.
HOST RESPONSE
Initially, the host responds to plasmodial infection by activating nonspecific defense mechanisms. Splenic immunologic and filtrative clearance functions are augmented in malaria, and the removal of both parasitized and uninfected erythrocytes is accelerated. The spleen is able to remove damaged ring-form parasites and return the once-infected erythrocytes to the circulation, where their survival period is shortened. The parasitized cells escaping splenic removal are destroyed when the schizont ruptures. The material released induces the activation of macrophages and the release of proinflammatory cytokines, which cause fever and exert other pathologic effects. Temperatures of ≥40°C (104°F) damage mature parasites; in untreated infections, the effect of such temperatures is to further synchronize the parasitic cycle, with eventual production of the regular fever spikes and rigors that originally served to characterize the different malarias. These regular fever patterns (quotidian, daily; tertian, every 2 days; quartan, every 3 days) are seldom seen today in patients who receive prompt and effective antimalarial treatment.
The geographic distributions of sickle cell disease, hemoglobins C and E, hereditary ovalocytosis, the thalassemias, and glucose-6-phosphate dehydrogenase (G6PD) deficiency closely resemble that of falciparum malaria before the introduction of control measures. This similarity suggests that these genetic disorders confer protection against death from falciparum malaria. For example, HbA/S heterozygotes (sickle cell trait) have a sixfold reduction in the risk of dying from severe falciparum malaria. Hemoglobin S–containing RBCs impair parasite growth at low oxygen tensions, and P. falciparum–infected RBCs containing hemoglobins S and C exhibit reduced cytoadherence because of reduced surface presentation of the adhesin PfEMP1. Parasite multiplication in HbA/E heterozygotes is reduced at high parasite densities. In Melanesia, children with α-thalassemia appear to have more frequent malaria (both vivax and falciparum) in the early years of life, and this pattern of infection appears to protect them against severe disease. In Melanesian ovalocytosis, rigid erythrocytes resist merozoite invasion, and the intraerythrocytic milieu is hostile.
Nonspecific host defense mechanisms stop the infection’s expansion, and the subsequent strain-specific immune response then controls the infection. Eventually, exposure to sufficient strains confers protection from high-level parasitemia and disease but not from infection. As a result of this state of infection without illness (premunition), asymptomatic parasitemia is common among adults and older children living in regions with stable and intense transmission (i.e., holo- or hyperendemic areas) and also in parts of low-transmission areas. Immunity is mainly specific for both the species and the strain of infecting malarial parasite. Both humoral immunity and cellular immunity are necessary for protection, but the mechanisms of each are incompletely understood (Fig. 248-1). Immune individuals have a polyclonal increase in serum levels of IgM, IgG, and IgA, although much of this antibody is unrelated to protection. Antibodies to a variety of parasitic antigens presumably act in concert to limit in vivo replication of the parasite. In the case of falciparum malaria, the most important of these antigens is the surface adhesin—the variant protein PfEMP1. Passively transferred IgG from immune adults has been shown to reduce levels of parasitemia in children. Passive transfer of maternal antibody contributes to the relative (but not complete) protection of infants from severe malaria in the first months of life. This complex immunity to disease declines when a person lives outside an endemic area for several months or longer.
Several factors retard the development of cellular immunity to malaria. These factors include the absence of major histocompatibility antigens on the surface of infected RBCs, which precludes direct T cell recognition; malaria antigen–specific immune unresponsiveness; and the enormous strain diversity of malarial parasites, along with the ability of the parasites to express variant immunodominant antigens on the erythrocyte surface that change during the course of infection. Parasites may persist in the blood for months or years (or, in the case of P. malariae, for decades) if treatment is not given. The complexity of the immune response in malaria, the sophistication of the parasites’ evasion mechanisms, and the lack of a good in vitro correlate with clinical immunity have all slowed progress toward an effective vaccine.
CLINICAL FEATURES
Malaria is a very common cause of fever in tropical countries. The first symptoms of malaria are nonspecific; the lack of a sense of well-being, headache, fatigue, abdominal discomfort, and muscle aches followed by fever are all similar to the symptoms of a minor viral illness. In some instances, a prominence of headache, chest pain, abdominal pain, cough, arthralgia, myalgia, or diarrhea may suggest another diagnosis. Although headache may be severe in malaria, the neck stiffness and photophobia seen in meningitis do not occur. While myalgia may be prominent, it is not usually as severe as in dengue fever, and the muscles are not tender as in leptospirosis or typhus. Nausea, vomiting, and orthostatic hypotension are common. The classic malarial paroxysms, in which fever spikes, chills, and rigors occur at regular intervals, are relatively unusual and suggest infection with P. vivax or P. ovale. The fever is usually irregular at first (that of falciparum malaria may never become regular); the temperature of nonimmune individuals and children often rises above 40°C (104°F) in conjunction with tachycardia and sometimes delirium. Although childhood febrile convulsions may occur with any of the malarias, generalized seizures are specifically associated with falciparum malaria and may herald the development of encephalopathy (cerebral malaria). Many clinical abnormalities have been described in acute malaria, but most patients with uncomplicated infections have few abnormal physical findings other than fever, malaise, mild anemia, and (in some cases) a palpable spleen. Anemia is common among young children living in areas with stable transmission, particularly where resistance has compromised the efficacy of antimalarial drugs. In nonimmune individuals with acute malaria, the spleen takes several days to become palpable, but splenic enlargement is found in a high proportion of otherwise healthy individuals in malaria-endemic areas and reflects repeated infections. Slight enlargement of the liver is also common, particularly among young children. Mild jaundice is common among adults; it may develop in patients with otherwise uncomplicated malaria and usually resolves over 1–3 weeks. Malaria is not associated with a rash like those seen in meningococcal septicemia, typhus, enteric fever, viral exanthems, and drug reactions. Petechial hemorrhages in the skin or mucous membranes—features of viral hemorrhagic fevers and leptospirosis—develop only very rarely in severe falciparum malaria.
SEVERE FALCIPARUM MALARIA
Appropriately and promptly treated, uncomplicated falciparum malaria (i.e., the patient can swallow medicines and food) carries a mortality rate of <0.1%. However, once vital-organ dysfunction occurs or the total proportion of erythrocytes infected increases to >2% (a level corresponding to >1012 parasites in an adult), mortality risk rises steeply. The major manifestations of severe falciparum malaria are shown in Table 248-2, and features indicating a poor prognosis are listed in Table 248-3.
MANIFESTATIONS OF SEVERE FALCIPARUM MALARIA |
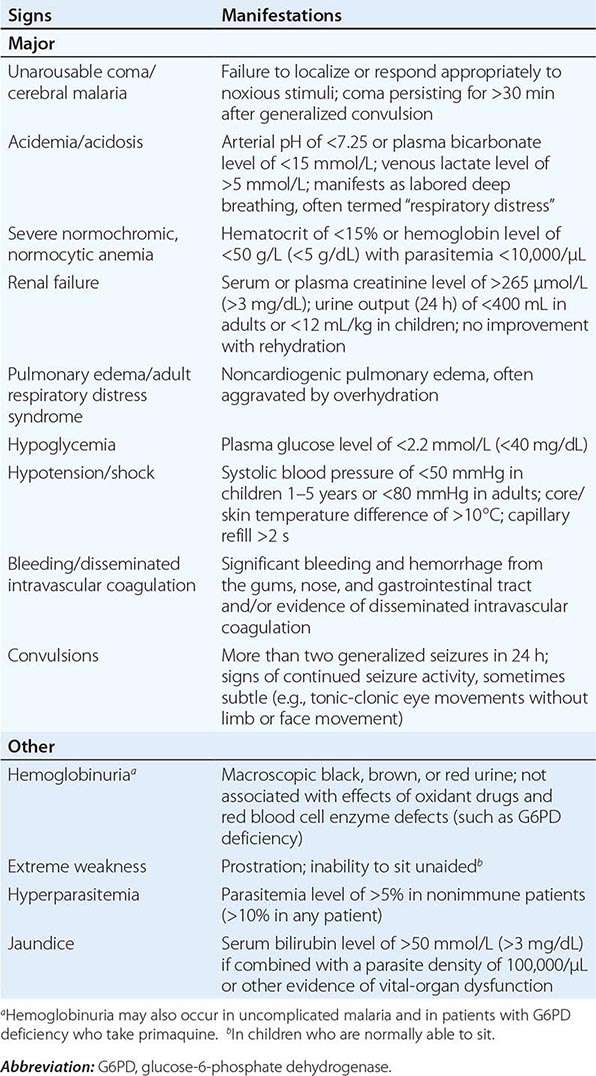
FEATURES INDICATING A POOR PROGNOSIS IN SEVERE FALCIPARUM MALARIA |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; PCV, packed cell volume.
Cerebral Malaria Coma is a characteristic and ominous feature of falciparum malaria and, despite treatment, is associated with death rates of ~20% among adults and 15% among children. Any obtundation, delirium, or abnormal behavior should be taken very seriously. The onset may be gradual or sudden following a convulsion.
Cerebral malaria manifests as diffuse symmetric encephalopathy; focal neurologic signs are unusual. Although some passive resistance to head flexion may be detected, signs of meningeal irritation are absent. The eyes may be divergent and a pout reflex is common, but other primitive reflexes are usually absent. The corneal reflexes are preserved, except in deep coma. Muscle tone may be either increased or decreased. The tendon reflexes are variable, and the plantar reflexes may be flexor or extensor; the abdominal and cremasteric reflexes are absent. Flexor or extensor posturing may be seen. On routine funduscopy, ~15% of patients have retinal hemorrhages; with pupillary dilation and indirect ophthalmoscopy, this figure increases to 30–40%. Other funduscopic abnormalities (Fig. 248-3) include discrete spots of retinal opacification (30–60%), papilledema (8% among children, rare among adults), cotton wool spots (<5%), and decolorization of a retinal vessel or segment of vessel (occasional cases). Convulsions, usually generalized and often repeated, occur in ~10% of adults and up to 50% of children with cerebral malaria. More covert seizure activity also is common, particularly among children, and may manifest as repetitive tonic-clonic eye movements or even hypersalivation. Whereas adults rarely (i.e., in <3% of cases) suffer neurologic sequelae, ~10% of children surviving cerebral malaria—especially those with hypoglycemia, severe anemia, repeated seizures, and deep coma—have residual neurologic deficits when they regain consciousness; hemiplegia, cerebral palsy, cortical blindness, deafness, and impaired cognition have been reported. The majority of these deficits improve markedly or resolve completely within 6 months. However, the prevalence of some other deficits increases over time; ~10% of children surviving cerebral malaria have a persistent language deficit. There may also be deficits in learning, planning and executive functions, attention, memory, and nonverbal functioning. The incidence of epilepsy is increased and life expectancy decreased among these children.
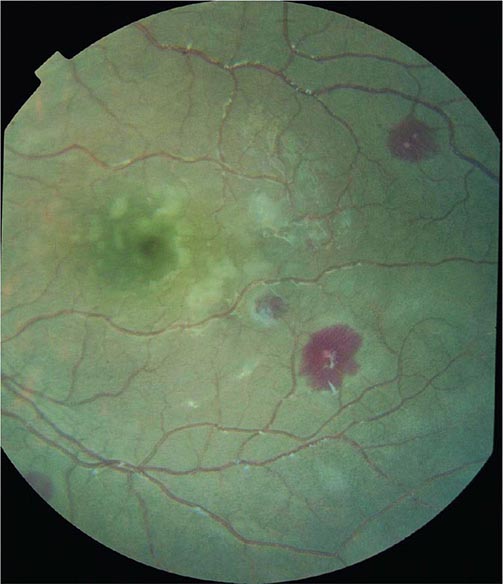
FIGURE 248-3 The eye in cerebral malaria: perimacular whitening and pale-centered retinal hemorrhages. (Courtesy of N. Beare, T. Taylor, S. Harding, S. Lewallen, and M. Molyneux; with permission.)
Hypoglycemia Hypoglycemia, an important and common complication of severe malaria, is associated with a poor prognosis and is particularly problematic in children and pregnant women. Hypoglycemia in malaria results from a failure of hepatic gluconeogenesis and an increase in the consumption of glucose by both the host and, to a much lesser extent, the malaria parasites. To compound the situation, quinine, which is still widely used for the treatment of both severe and uncomplicated falciparum malaria, is a powerful stimulant of pancreatic insulin secretion. Hyperinsulinemic hypoglycemia is especially troublesome in pregnant women receiving quinine treatment. In severe disease, the clinical diagnosis of hypoglycemia is difficult: the usual physical signs (sweating, gooseflesh, tachycardia) are absent, and the neurologic impairment caused by hypoglycemia cannot be distinguished from that caused by malaria.
Acidosis Acidosis, an important cause of death from severe malaria, results from accumulation of organic acids. Hyperlactatemia commonly coexists with hypoglycemia. In adults, coexisting renal impairment often compounds the acidosis; in children, ketoacidosis also may contribute. Other, still-unidentified organic acids are major contributors to acidosis. Acidotic breathing, sometimes called “respiratory distress,” is a sign of poor prognosis. It is followed often by circulatory failure refractory to volume expansion or inotropic drug treatment and ultimately by respiratory arrest. The plasma concentrations of bicarbonate or lactate are the best biochemical prognosticators in severe malaria. Hypovolemia is not a major contributor to acidosis. Lactic acidosis is caused by the combination of anaerobic glycolysis in tissues where sequestered parasites interfere with microcirculatory flow, lactate production by the parasites, and a failure of hepatic and renal lactate clearance. The prognosis of severe acidosis is poor.
Noncardiogenic Pulmonary Edema Adults with severe falciparum malaria may develop noncardiogenic pulmonary edema even after several days of antimalarial therapy. The pathogenesis of this variant of the adult respiratory distress syndrome is unclear. The mortality rate is >80%. This condition can be aggravated by overly vigorous administration of IV fluid. Noncardiogenic pulmonary edema can also develop in otherwise uncomplicated vivax malaria, where recovery is usual.
Renal Impairment Acute kidney injury is common in severe falciparum malaria, but oliguric renal failure is rare among children. The pathogenesis of renal failure is unclear but may be related to erythrocyte sequestration and agglutination interfering with renal microcirculatory flow and metabolism. Clinically and pathologically, this syndrome manifests as acute tubular necrosis. Renal cortical necrosis never develops. Acute renal failure may occur simultaneously with other vital-organ dysfunction (in which case the mortality risk is high) or may progress as other disease manifestations resolve. In survivors, urine flow resumes in a median of 4 days, and serum creatinine levels return to normal in a mean of 17 days (Chap. 334). Early dialysis or hemofiltration considerably enhances the likelihood of a patient’s survival, particularly in acute hypercatabolic renal failure.
Hematologic Abnormalities Anemia results from accelerated RBC removal by the spleen, obligatory RBC destruction at parasite schizogony, and ineffective erythropoiesis. In severe malaria, both infected and uninfected RBCs show reduced deformability, which correlates with prognosis and development of anemia. Splenic clearance of all RBCs is increased. In nonimmune individuals and in areas with unstable transmission, anemia can develop rapidly and transfusion is often required. As a consequence of repeated malarial infections, children in many areas of Africa and on the island of New Guinea may develop severe anemia resulting from both shortened survival of uninfected RBCs and marked dyserythropoiesis. Anemia is a common consequence of antimalarial drug resistance, which results in repeated or continued infection.
Slight coagulation abnormalities are common in falciparum malaria, and mild thrombocytopenia is usual (a normal platelet count should raise questions about the diagnosis of malaria). Of patients with severe malaria, <5% have significant bleeding with evidence of disseminated intravascular coagulation. Hematemesis from stress ulceration or acute gastric erosions also may occur rarely.
Liver Dysfunction Mild hemolytic jaundice is common in malaria. Severe jaundice is associated with P. falciparum infections; is more common among adults than among children; and results from hemolysis, hepatocyte injury, and cholestasis. When accompanied by other vital-organ dysfunction (often renal impairment), liver dysfunction carries a poor prognosis. Hepatic dysfunction contributes to hypoglycemia, lactic acidosis, and impaired drug metabolism. Occasional patients with falciparum malaria may develop deep jaundice (with hemolytic, hepatic, and cholestatic components) without evidence of other vital-organ dysfunction, in which case the prognosis is good.
Other Complications HIV/AIDS and malnutrition predispose to more severe malaria in nonimmune individuals; malaria anemia is worsened by concurrent infections with intestinal helminths, hookworm in particular. Septicemia may complicate severe malaria, particularly in children. Differentiating severe malaria from sepsis with incidental parasitemia in childhood is very difficult. In endemic areas, Salmonella bacteremia has been associated specifically with P. falciparum infections. Chest infections and catheter-induced urinary tract infections are common among patients who are unconscious for >3 days. Aspiration pneumonia may follow generalized convulsions. The frequencies of complications of severe falciparum malaria are summarized in Table 248-4.
RELATIVE INCIDENCE OF SEVERE COMPLICATIONS OF FALCIPARUM MALARIA |
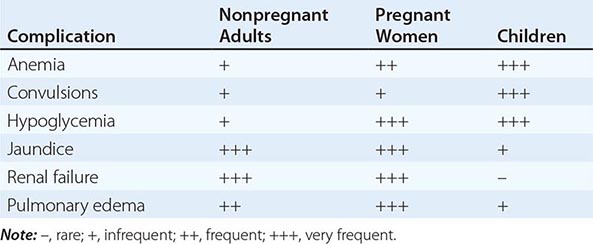
MALARIA IN PREGNANCY
Malaria in early pregnancy causes abortion. In areas of high malaria transmission, falciparum malaria in primi- and secundigravid women is associated with low birth weight (average reduction, ~170 g) and consequently increased infant mortality rates. In general, infected mothers in areas of stable transmission remain asymptomatic despite intense accumulation of parasitized erythrocytes in the placental microcirculation. Maternal HIV infection predisposes pregnant women to more frequent and higher-density malaria infections, predisposes their newborns to congenital malarial infection, and exacerbates the reduction in birth weight associated with malaria.
In areas with unstable transmission of malaria, pregnant women are prone to severe infections and are particularly vulnerable to high parasitemias with anemia, hypoglycemia, and acute pulmonary edema. Fetal distress, premature labor, and stillbirth or low birth weight are common results. Fetal death is usual in severe malaria. Congenital malaria occurs in <5% of newborns whose mothers are infected; its frequency and the level of parasitemia are related directly to the parasite density in maternal blood and in the placenta. P. vivax malaria in pregnancy is also associated with a reduction in birth weight (average, 110 g), but, in contrast to the situation in falciparum malaria, this effect is more pronounced in multigravid than in primigravid women. About 350,000 women die in childbirth yearly, with most deaths occurring in low-income countries; maternal death from hemorrhage at childbirth is correlated with malaria-induced anemia.
MALARIA IN CHILDREN
Most of the 660,000 persons who die of falciparum malaria each year are young African children. Convulsions, coma, hypoglycemia, metabolic acidosis, and severe anemia are relatively common among children with severe malaria, whereas deep jaundice, oliguric acute kidney injury, and acute pulmonary edema are unusual. Severely anemic children may present with labored deep breathing, which in the past has been attributed incorrectly to “anemic congestive cardiac failure” but in fact is usually caused by metabolic acidosis, often compounded by hypovolemia. In general, children tolerate antimalarial drugs well and respond rapidly to treatment.
TRANSFUSION MALARIA
Malaria can be transmitted by blood transfusion, needle-stick injury, sharing of needles by infected injection drug users, or organ transplantation. The incubation period in these settings is often short because there is no preerythrocytic stage of development. The clinical features and management of these cases are the same as for naturally acquired infections. Radical chemotherapy with primaquine is unnecessary for transfusion-transmitted P. vivax and P. ovale infections.
CHRONIC COMPLICATIONS OF MALARIA
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


