Alveolar Rhabdomyosarcoma
Khin Thway, BSc, MBBS, FRCPath
Key Facts
Terminology
High-grade malignant round cell neoplasm showing partial differentiation towards skeletal muscle
Etiology/Pathogenesis
Characteristic PAX-FOXO1 balanced translocations in most
t(2;13)(q35;q14) (majority)
t(1;13)(p36;q14) in 10-15%
Clinical Issues
Accounts for approximately 30% of rhabdomyosarcomas
Predominantly adolescents and young adults
Sites: Deep soft tissue of extremities, head and neck, and trunk
Can present with widespread dissemination
Tend to be high-stage lesions at presentation
Microscopic Pathology
Poorly differentiated round cells
Hyperchromatic nuclei, scanty cytoplasm
Alveolar-like spaces formed by central loss of cohesion
Multinucleate giant tumor cells in some cases
Fibrovascular septa separate nests
Rhabdomyoblasts can be present
Solid variant
Lacks alveolar architecture
Mixed alveolar and embryonal rhabdomyosarcoma
Behavior and classification as ARMS
Desmin usually strong and diffuse
Myogenin and MYOD1 are sensitive and specific for skeletal muscle differentiation
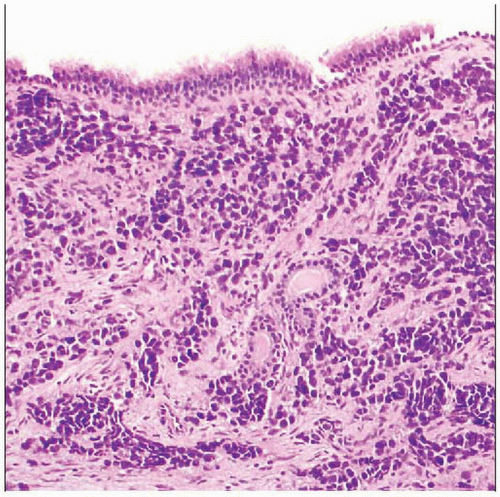 Alveolar rhabdomyosarcoma from the postnasal space is seen. Respiratory-type epithelium overlies fibrous tissue extensively infiltrated by sheets and nests of monomorphic round cell tumor. |
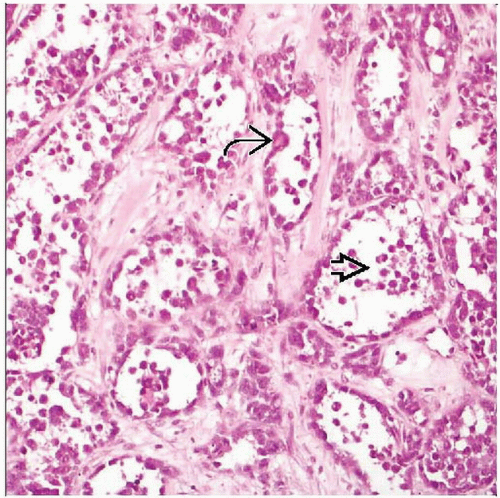 The tumor is composed of round cells with peripheral cellular preservation  , but central discohesion and necrosis , but central discohesion and necrosis  as well, somewhat resembling the appearance of pulmonary alveoli. as well, somewhat resembling the appearance of pulmonary alveoli. |
TERMINOLOGY
Abbreviations
Alveolar rhabdomyosarcoma (ARMS)
Definitions
High-grade malignant round cell neoplasm characterized by recurrent chromosomal translocations showing variable differentiation toward skeletal muscle
ETIOLOGY/PATHOGENESIS
Genetic Events
Characteristic balanced translocations
Produce chimeric fusion proteins
These act as aberrant transcription factors
Regulate expression of specific target genes
Target cell/cell of origin still unknown
CLINICAL ISSUES
Epidemiology
Incidence
Accounts for approximately 30% of rhabdomyosarcomas
2nd most common rhabdomyosarcoma
After embryonal RMS
Age
Predominantly adolescents and young adults
Peak incidence: 10-25 years
Rare in adults > 45 years
Congenital in small number of cases
Gender
M = F
Ethnicity
No ethnic or geographical predilections
Site
Deep soft tissue
Most common in extremities
Head and neck
Trunk
Pelvis
Retroperitoneum
Perineum
Presentation
Suddenly enlarging mass
Local symptoms pertaining to site of origin
Proptosis or cranial nerve deficits (head and neck)
Paresthesia or paresis (paraspinal areas of trunk)
Can present with widespread dissemination
Lymphadenopathy or marrow infiltration
Rarely may present without obvious primary
Deep mass
Treatment
Multimodality approach
RMS is sensitive to both chemotherapy and radiation therapy
Systemic chemotherapy
In conjunction with surgery, radiation therapy, or both modalities
Radiotherapy
To maximize local control
Surgery
Excise primary tumor when possible, without major functional or cosmetic defects
Complete resection often difficult or impossible
Prognosis
Tend to be high-stage lesions at presentation
Prognosis of ARMS significantly worse than for embryonal RMS
Accurate distinction between RMS subtypes is therefore crucial for appropriate management
MACROSCOPIC FEATURES
General Features
Fleshy mass
Infiltrative margins
Tan cut surface
Hemorrhage
Necrosis
MICROSCOPIC PATHOLOGY
Histologic Features
Poorly differentiated round cells
Medium size
Hyperchromatic nuclei
Scanty cytoplasm
Sheets and nests
Alveolar-like spaces formed by central loss of cohesion
Central cells poorly preserved and necrotic
Many appear freely floating
Fibrovascular septa separate nests
Rare clear cell appearance
Due to cytoplasmic glycogen
Vacuolated cells may be confused with lipoblasts
Rhabdomyoblasts can be present
Less frequent than in embryonal RMS
Multinucleate giant tumor cells in some cases
Peripheral or wreath-like nuclei
Very rarely atypical cells, similar to anaplastic variant of embryonal RMS
Tumor metastases often show alveolar pattern
Solid variant
Sheets and islands of densely packed tumor cells
Cytomorphology of typical ARMS
Lack alveolar pattern
Occasional small nests may be present
Foci of more typical ARMS may be seen on careful examination
More likely to be fusion(-) for PAX3/7-FOXO1
Very rarely atypical cells, similar to anaplastic variant of embryonal RMS
Mixed alveolar and embryonal rhabdomyosarcoma
Foci of embryonal morphology
Spindle cells or myxoid stroma
Behavior and classification as ARMS
Lymph Nodes
Nodal metastases may be presenting factor
Predominant Pattern/Injury Type
Neoplastic
Predominant Cell/Compartment Type
Mesenchymal, muscle, skeletal
Genetics
Characteristic balanced recurrent translocations
Majority of cases (80-85%) of ARMS
Translocations are not feature of other RMS subtypes
Fusion of 2 genes, each encoding transcription factors
FOXO1
Chromosome 13
Member of forkhead transcription factor family
PAX3 or PAX7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


