Chapter 4 Acute alveolar injury and repair
Acute repiratory distress syndrome
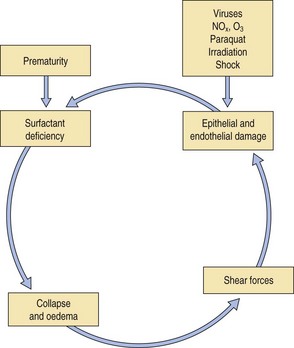
Acute alveolar injury may be caused by a wide range of pulmonary insults that at their most severe result in what is termed the acute respiratory distress syndrome (ARDS). This was formerly known as the adult respiratory distress syndrome,1–3 a term introduced to emphasise its similarity to that seen in premature babies suffering from the effects of deficient pulmonary surfactant production, i.e. the infantile respiratory distress syndrome (see p. 43), but following the recognition that the causes of the adult respiratory distress syndrome may also operate in children, ‘adult’ has given way to ‘acute’. The causes of the syndrome differ markedly in infants and adults but whatever the cause, a common cycle of events is initiated (Fig. 4.1) so that the end-result is the same regardless of the cause. It is a common condition, which carries a high mortality, about 50% overall. Estimations of incidence show considerable international variability but one American study reported it to be 79 per 100000 population.4
ARDS is a fulminant form of respiratory failure characterised by refractory hypoxaemia (Pao2/FIo2 < 200) and bilateral radiographic opacification in the absence of any evidence of an elevated left atrial pressure.5 These features indicate widespread alveolar collapse and exudation that cannot be attributed to left heart failure or other cause of pulmonary venous hypertension. There is generalised ground-glass opacification of the lungs, which is most pronounced in the dependent parts of the lungs. The opacification rapidly becomes increasingly dense until there is frank consolidation (Fig. 4.2).6 Air is then confined to the bronchi, which therefore appear black on plain radiographs where they stand out against the alveolar ‘white-out’ (so-called air bronchograms).7
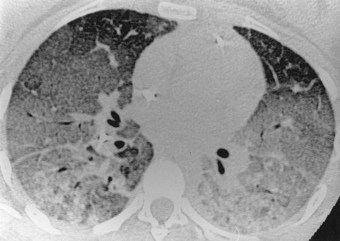
Functional studies confirm that little of the lung substance is ventilated. There is initially mild pulmonary hypertension but the pulmonary arterial constriction responsible for this is succeeded by vascular non-responsiveness so that the normal vasoconstrictor response to hypoxia is diminished. The consequent ventilation/perfusion mismatching aggravates the hypoxaemia. Other organs suffer both hypoxia and the effects of inflammatory mediators initiated by the pulmonary injury once these gain access to the general circulation. The end stages of ARDS are therefore frequently associated with multiple organ failure.8
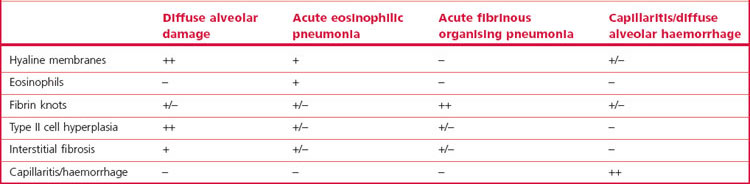
Despite the variety of causes, most patients with ARDS show the same pattern of pathological changes, one that is termed diffuse alveolar damage (DAD). Much of this chapter is devoted to this pattern of injury but occasionally patients presenting with ARDS show other changes when subjected to biopsy or coming to autopsy. These are listed in Box 4.1 and are considered in other chapters but their chief pathological features are compared with those of DAD in Table 4.1. Recognition of these other patterns may lead to a change in the patient’s management. In that they include infection it is essential that some of the specimen goes to the microbiology department. Lung biopsy is also undertaken to assess the reversibility of the changes.9,10 The process is potentially reversible in its exudative phase whereas widespread fibrosis with loss of the lung architecture is not.
Box 4.1 The pathological basis of the acute respiratory distress syndrome
Diffuse alveolar damage
DAD represents a non-specific pattern of acute alveolar injury caused by a variety of noxious agents.11–14 It is the chief pathological basis of ARDS. The pathological changes of DAD can be divided into the overlapping phases of exudation, regeneration and repair.15
Exudative phase
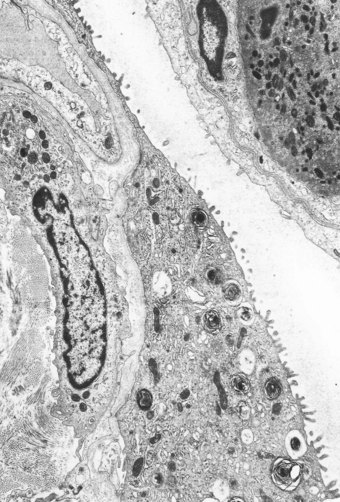
To facilitate gas exchange the alveolar wall is highly specialised in structure. Unfortunately this specialisation renders it susceptible to injury by a wide variety of agents. The principal cells of the air–blood barrier, the type I alveolar epithelial cell and the capillary endothelial cell are exceptionally thin (see Fig. 1.27, p. 16) and this makes them particularly vulnerable to non-specific damage. Injury to these two cells underlies the development of DAD. At an early stage of alveolar injury the type I epithelial cells show cytoplasmic blebbing, which is soon followed by necrosis resulting in denudation of the basement membrane (see Fig. 7.2.5, p. 376).16,17 Similar blebbing is seen in the alveolar capillary endothelium but denudation of the endothelial basement membrane is seldom observed, probably because of differences in the ways epithelial and endothelial cells regenerate (see below). The consequences of this damage include the escape of fibrin-rich exudates into the interstitial and air spaces, loss of the surface-active alveolar lining film and pulmonary collapse.
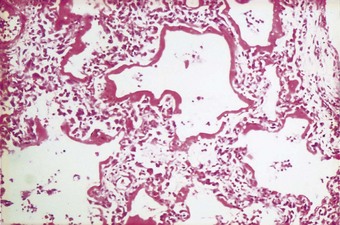
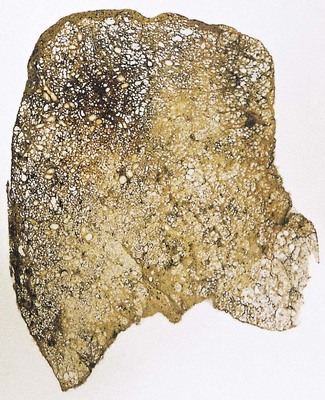
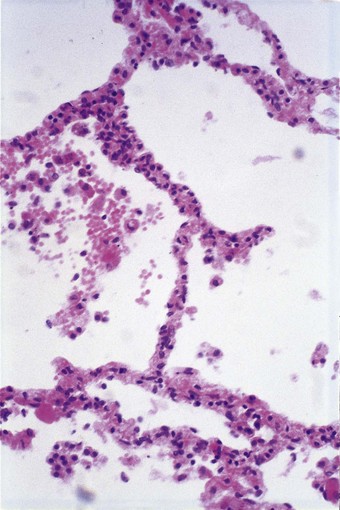
The exudative phase lasts about 1 week, during which the lungs are heavy, often weighing over 1 kg each, dark and airless. The changes are often patchy, with the dorsal and basal regions being most severely affected (Fig. 4.3).18–20 Slicing shows that they are wet, the cut surface exuding blood or heavily blood-stained watery fluid. Microscopically there is widespread collapse, intense congestion of the capillaries, interstitial oedema and distension of the lymphatics, a pattern that is sometimes known as congestive atelectasis (Fig. 4.4). Alternatively, there may be haemorrhagic oedema (Fig. 4.5). At the air–tissue interface, which in these collapsed lungs is at the respiratory bronchiole or alveolar duct level, respiratory movements compact a fibrin-rich exudate mixed with necrotic epithelial debris into a thin layer that covers an otherwise denuded epithelial basement membrane (Fig. 4.6), leading to the formation of hyaline membranes (Fig. 4.7)11–13,21,22: these are identical to those that paediatric pathologists recognise as the hallmark of the infantile respiratory distress syndrome (compare Fig. 4.3 with Fig. 2.8, p. 44). Similar changes are also seen in acute eosinophilic pneumonia but here the hyaline membranes contain eosinophils, possibly in small focal collections that are easily overlooked (see p. 462).
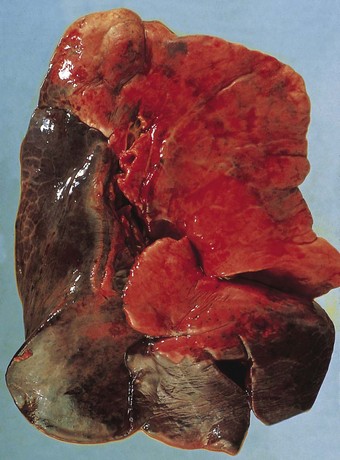
Figure 4.3 Shock lung. The lower lobe shows large irregular areas of collapse and congestion.
(Reproduced from Corrin (1980)18 by permission of the editor of the Journal of Clinical Pathology.)
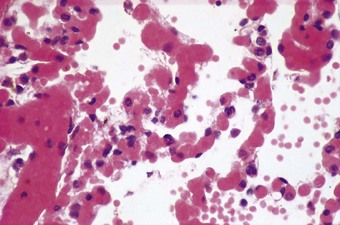
Figure 4.4 Congestive atelectasis in septic shock. There is severe capillary congestion and alveolar collapse.
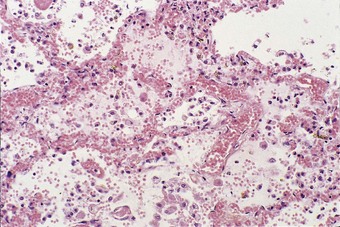
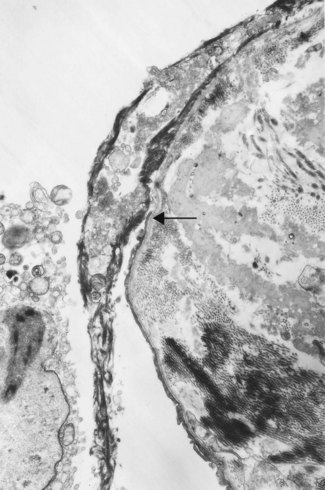
The congested alveolar capillaries sometimes contain increased numbers of platelets or neutrophil leukocytes. This selective sequestration of formed blood elements in the microvasculature of the lungs is particularly noticeable in shock, and is considered in more detail under that heading below.
Regenerative phase
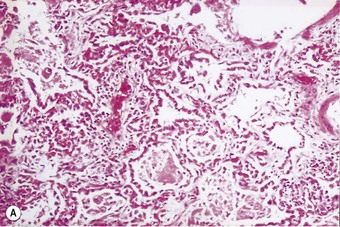
The regenerative (or proliferative) phase becomes prominent 1–2 weeks after the initial injury. It involves proliferation of both epithelial and connective tissue cells. The stem cell concerned in epithelial regeneration is the type II alveolar epithelial cell.23,24 These cells first proliferate and then differentiate into type I cells, thereby re-epithelialising the denuded basement membranes. The dividing type II cells form a simple cuboidal epithelium (Fig. 4.8), or the alveoli may be lined by plump pleomorphic spindle cells that represent cell forms intermediate between types II and I epithelial cells (Figs 4.9 and 4.10). Eosinophilic cytoplasmic inclusions indicative of cell damage (see Fig. 7.1.25, p. 348) are often present,25,26 and sometimes there is squamous metaplasia instead of orderly differentiation into type I cells, a change that the unwary pathologist may mistake for neoplasia.27
Figure 4.8 Diffuse alveolar damage, regenerative phase. (A) The alveoli have a simple cuboidal epithelial lining.
(Courtesy of Dr D Melcher, Brighton, UK.)
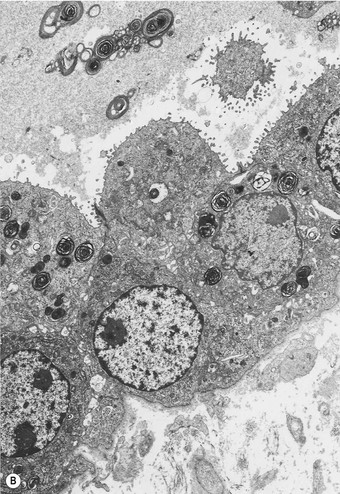
The regenerating epithelium usually grows beneath the exudates lining the denuded basement membrane, casting the hyaline membranes off into the air space (Fig. 4.11), but it may grow over them so that they are incorporated into the interstitium (Figs 4.12 and 4.13),11–13 where their subsequent organisation contributes to the fibrosis.22 The regenerating epithelial cells may also bridge the mouths of collapsed alveoli so that these air spaces never re-expand and there is permanent shrinkage of the lung, a process termed atelectatic induration (Figs 4.14 and 4.15).28–30
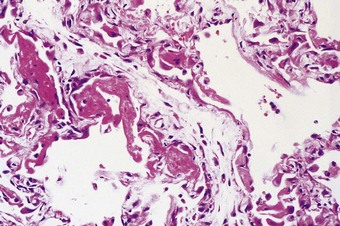
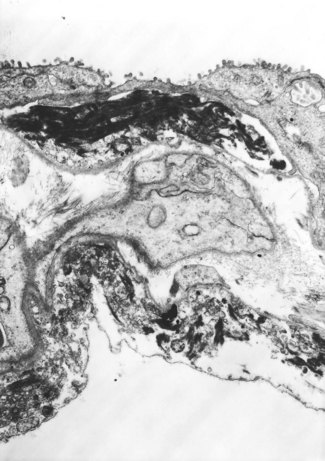
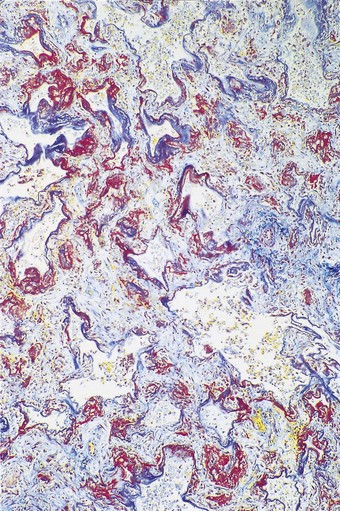
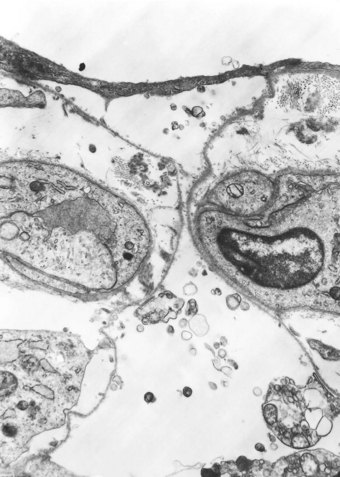
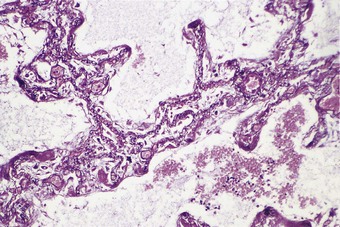
In contrast to the type I epithelial cells, which have no regenerative powers and are replaced by differentiation of proliferating type II cells, endothelial cells are replaced by lateral spread of their own kind. An effete endothelial cell is first undermined by its healthy neighbours and only cast off when these have completely covered the basement membrane.31 Therefore, although segments of bare basement membrane have been described on the vascular side of the air–blood barrier,32,33 they are not seen to the same extent as on the epithelial side. Nevertheless, thrombosis may complicate such endothelial damage21,32 and subsequent organisation of such thrombi is probably responsible for some of the vascular remodelling that is seen in the repair phase of DAD. This remodelling consists of fibrocellular intimal thickening that narrows the lumen of small vessels throughout the lung and can be visualised as decreased background filling on postmortem arteriograms.34
Repair phase
If healing is by repair, interstitial connective tissue cells proliferate and, as in any scarring, myofibroblasts are involved at an early stage.35 Whilst myofibroblast contracture is beneficial when it promotes early closure of an open wound, in the lungs it largely results in harmful distortion of the bronchioloalveolar architecture and shrinkage of the lungs. Possible sources of the proliferating interstitial connective tissues include resting interstitial connective tissue cells, the bone marrow and the alveolar epithelium. Epithelial–mesenchymal transition is being increasingly recognised throughout the body and in the lung in conditions such as idiopathic pulmonary fibrosis, asthma and obliterative bronchiolitis.36–38
Whatever the source of the proliferating connective tissue cells, those with a fibroblastic phenotype lay down collagen, leading to the development of interstitial fibrosis.39,40 Interactions between fibroblasts and the alveolar epithelium through gaps in the basement membrane have been described,41–45 suggesting that the regenerating epithelial cells play a further role in the underlying process of fibrosis. It is known that these cells synthesise fibrogenic cytokines such as tumour necrosis factor-α; the secretion of tumour necrosis factor-α into the underlying connective tissue would further promote interstitial fibrosis.46,47
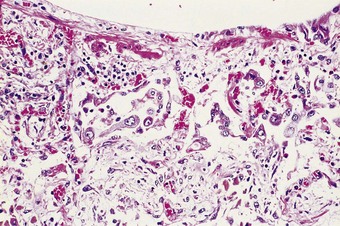
Fibroblasts also migrate into the alveolar exudates through defects in the epithelial basement membrane to lay down collagen within the hyaline membranes.48,49 As epithelial cells grow over the newly formed connective tissue, a new basement membrane is formed, thereby incorporating the collagen into the interstitium,49 a process known as fibrosis by accretion. Involvement of the alveolar ducts results in these structures being lined by a ring of granulation tissue (Fig. 4.16). Less frequently, the organised exudates retain a predominantly intra-alveolar position, resulting in loose buds of granulation tissue similar to those seen in organising pneumonia due to other causes (see Box 6.2.1, p. 309). However, here they are more widespread and there is more prominent type II cell hyperplasia, interstitial fibroblastic proliferation and interstitial inflammation (Fig. 4.17). Nevertheless, such an organising pneumonia pattern is worth recognising as it carries a better prognosis than interstitial fibrosis.50 It is claimed that this intra-alveolar pattern of repair is particularly found when generalised sepsis is the cause of the initial damage whereas interstitial fibrosis is more characteristic of injury caused by cytotoxic drugs and the idiopathic cases.51
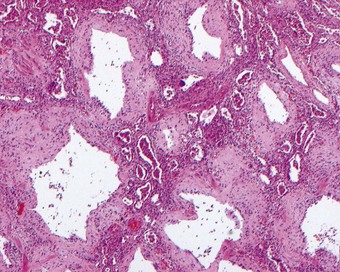
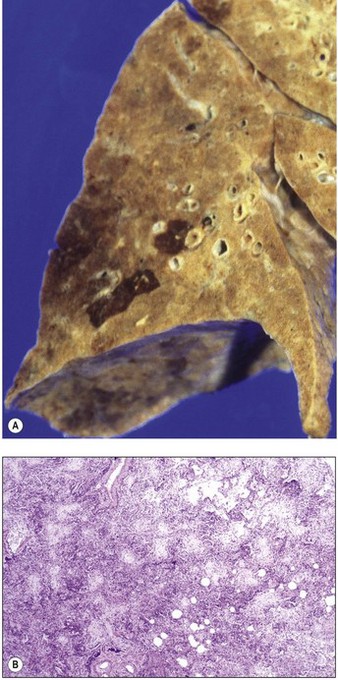
An increase in lung collagen can be detected in patients with ARDS who survive longer than 14 days and this progressively increases with the duration of the disease.52 The identification of pulmonary fibrosis on transbronchial biopsy is closely related to mortality.53 Fibrosis can be well established by 2 weeks, at which time the lungs may be contracted and firm with a fine sponge-like pattern on their cut surfaces, representing bronchiolectasis and irregular microcystic distortion of the alveolar architecture (Figs 4.18 and 4.19).54–57 The changes are similar to those seen in any fibrotic process but are reached remarkably quickly (see Fig. 4.19).22 However, at this early stage the fibrosis differs from that seen in chronic fibrosis in being more cellular and less collagenous: extensive fibroblast proliferation is evident. With small air cysts alternating with solid areas of fibrosis and foci of squamous metaplasia there is a resemblance to the bronchopulmonary dysplasia seen in the late stages of the infantile respiratory distress syndrome.55,56 In survivors the granulation tissue undergoes progressive collagenisation and such patients may suffer from debilitating fibrotic lung disease. The pattern is then that of fibrotic non-specific interstitial pneumonia,58 making it important to determine whether there has been an episode of acute lung injury in such patients.
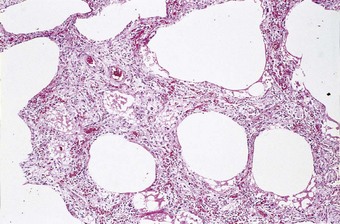
Figure 4.18 Diffuse alveolar damage, repair phase. The repair process has resulted in interstitial fibrosis.
Causes
The causes of DAD are quite diverse (Box 4.2). So too are the pathways by which the injurious agents reach the lungs. Some enter the lungs directly via the airways, e.g. oxygen in high concentrations, poisonous gases such as phosgene and metallic fumes such as those of mercury and cadmium. Other agents responsible for DAD penetrate the chest wall to damage the lungs (e.g. ionising radiation) and some reach the lungs via the blood stream, having been ingested or injected (e.g. paraquat and cytotoxic chemotherapeutic agents). The blood stream also conveys many of the endogenous factors that underlie the DAD of shock. In numerical terms, septic shock is the most important cause of DAD.59,60
Multiple causes may operate in one patient. For example, trauma may be combined with blood loss, fat embolism and sepsis, whilst therapeutic efforts to correct these may themselves be hazardous. The transfusion of stored blood is not without danger, whilst to prevent hypoxaemia, damaged lungs that require rest often have to be forcibly ventilated and subjected to injurious concentrations of oxygen, although it is known that this can only aggravate the injury to the lungs.22 The damaged lung also appears to be unduly susceptible to infection,61 partly because of impaired neutrophil migration into the air spaces.62 It is therefore common in clinical practice for the lungs to be subjected to several injurious agents and since these all contribute to a non-specific pattern of disease it may be difficult for the pathologist to distinguish the initiating factor from the effects of treatment. Consideration of events in the intensive care unit is essential in these circumstances. Many of the causes of DAD listed in Box 4.2 are dealt with elsewhere, leaving only a few to be considered here.
Shock
Shock is a state of prolonged hypotension, generally attributable to trauma, hypovolaemia, cardiac failure, sepsis or anaphylaxis.63,64 The hypotension leads to inadequate tissue perfusion and, if this is not corrected, multiorgan failure is inevitable. At necropsy, the lungs are the organs most commonly affected.65–67
Severe pulmonary injury was well described in patients suffering from shock in the Second World War68 but it was not until the war in Vietnam that the importance of respiratory failure as a complication of shock was fully appreciated.69,70 By this time there had been major improvements in medical care. Casualties could be transported rapidly by helicopter to well-equipped field hospitals where intensive care with mechanical ventilatory support was available. Despite this, injured patients often developed fatal respiratory insufficiency, typically after an interval of between 48 and 72 hours. A number of terms graphically described this syndrome – ‘shock lung’, ‘posttraumatic respiratory insufficiency’, ‘traumatic wet lung’ and ‘Da Nang lung’. Pathological examination showed congestive atelectasis or haemorrhagic oedema proceeding to fully developed DAD, as described above.
The pathogenesis of ‘shock lung’ is complex and requires special consideration. In hypovolaemic and cardiogenic shock, compensatory mechanisms such as peripheral vasoconstriction initially maintain cerebral oxygenation, but if the underlying cause is untreated, there follows a state of decompensation characterised by vascular unresponsiveness: vasodilatation develops, the blood pressure plummets and there is widespread hypoxic cell death. Anaphylactic and septic shock are characterised from the outset by such vasodilatation, which is caused by a variety of mediators that are released from inflammatory and other cells.71 The identification of the same mediators in experimental hypovolaemia72 suggests that the pathogenesis of shock may be similar regardless of the cause.
In numerical terms, septic shock is the most important cause of diffuse damage.59,60 It represents the culmination of a clinicopathological continuum of infection-driven sepsis syndromes:
These terms have replaced ‘septicaemia’, which proved difficult to define and has now been abandoned. The multiorgan dysfunction is due to inflammatory cytokines released into the circulation from the site of infection, the common sites of which are, in decreasing frequency, lung, blood, intestine and peritoneum, urinary tract and surgical wounds. Gram-positive infections are slightly commoner than Gram-negative, which are slightly commoner than polymicrobial. The role of lipopolysaccharide derived from the cell walls of Gram-negative bacteria has been particularly well studied in the pathogenesis of septic shock.73–77 The many effects of this endotoxin include the release of tumour necrosis factor from monocytes, macrophages and polymorphonuclear leukocytes,74–76,78,79 the production of cytokines such as interleukin-1 and interferon-γ that act synergistically with tumour necrosis factor,80 the widespread induction of nitric oxide synthesis81–83 and the activation of both the coagulation and complement cascades.84 Exotoxins released by Gram-positive bacteria appear to act similarly. Some of the most fulminant forms of shock are seen with group A streptococci causing puerperal sepsis and necrotising fasciitis, meningococci, Staphylococcus aureus related to tampon retention and a meticillin-resistant strain that secretes an exotoxin known as Panton–Valentine leukocidin.85
Tumour necrosis factor has been demonstrated on the luminal surface of pulmonary endothelium in endotoxin-induced shock.86 It causes vascular smooth muscle to relax but this action is reduced if the endothelium is removed,87 indicating that tumour necrosis factor-induced vasodilatation is partially dependent upon the integrity of the endothelium. A factor that causes vascular dilatation has been detected as coming from the vascular endothelium and acting on the medial muscle coat of the vessel. At first termed endothelium-derived relaxing factor, this factor is now known to be nitric oxide, a remarkably simple chemical that has long been recognised to be poisonous.88 Fortunately, its half-life in the vessel wall is very short, timed in seconds rather than minutes. The enzyme responsible for its production (from L-arginine) is nitric oxide synthase, which is found in endothelium and can be induced in the vascular medial smooth muscle. Both the constitutive and inducible forms of the enzyme are activated by bacterial lipopolysaccharide and it therefore seems likely that in septic shock bacterial products act directly on the vessel wall resulting in the production of excess amounts of nitric oxide. Even momentarily increased levels of nitric oxide might be expected to cause arterial dilatation and hence capillary congestion. It would appear that in septic shock circulating bacterial products such as lipopolysaccharide cause vascular dilatation and possibly increased permeability by direct action on the blood vessels and indirectly through the induction of nitric oxide synthase and the release of the cytokines mentioned in the preceding paragraph. Pulmonary endothelial upregulation is indicated by widespread expression of intercellular adhesion molecule-1 (ICAM-1: CD54) in septic shock. Indeed, positive immunostaining for CD54 is proving to be a useful marker of shock.89
Nitric oxide is a powerful vasodilator but it also mediates many other processes throughout the body. In host defence nitric oxide plays a very different and more aggressive role. It enables macrophages to generate free oxygen radicals, the principal means by which these cells eliminate both bacteria and cancer cells, but which, if not inactivated, also damage healthy host cells.90 Macrophages release much more nitric oxide than endothelial cells but, as in the blood vessels, the amounts released are generally inactivated within seconds. However, overwhelming bacterial infections result in the release of very large amounts of nitric oxide and the overproduction of toxic oxygen radicals. Although the oxygen radicals are countered by the protective action of enzymes such as superoxide dismutase,91 their excessive release results in oxidation of lipids and protein sulphydryl groups, and DNA damage.92 Damaged cell membrane phospholipids release free arachidonic acid which in turn is degraded to produce leukotrienes, such as prostaglandins and thromboxane, that are capable of altering vessel calibre and permeability.
The direct vasodilatory action of nitric oxide and the toxic action of the free oxygen radicals that nitric oxide generates account for most of the pathological features of shock but there are other processes in the microvasculature of the lung that contribute to the pulmonary damage. The vascular engorgement characteristic of shock lung is occasionally accompanied by sequestration of neutrophil polymorphonuclear leukocytes in the pulmonary microvasculature (Figs 4.20 and 4.21).18,18,93,94 Acting through the complement cascade,84,95,96 endotoxin activates these cells within the systemic circulation so that they lose their normal deformability and aggregate into microemboli, with the result that they cannot traverse the alveolar capillaries.73,97,98 Their arrest there is promoted by activated endothelial intercellular adhesion molecules.99–103 The unique position of the pulmonary capillaries in the circulation is probably responsible for the lungs being the organs most severely affected in shock.65–67 Trapped in the alveolar capillaries, activated neutrophils damage the alveolar wall by producing reactive oxygen radicals and releasing enzymes such as elastase, collagenase and cathepsins that are able to degrade protein constituents of the wall.104–110 Impairment of neutrophil migration into the air spaces has been referred to above.62
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


