Caused by mutations in SERPINA1 gene
•
Deficiency is characterized by emphysema and chronic liver disease
Etiology/Pathogenesis
•
Mutations result in defective secretion of molecule, accumulation in hepatocytes, and decreased serum A1AT

Specific mechanism of hepatocyte injury is unknown
•
Most common deficiency alleles are PiS and PiZ

PiZZ phenotype accounts for most cases of severe A1AT deficiency and virtually all cases of liver disease
Clinical Issues
•
Most common in Caucasians of Northern European ancestry
•
Liver disease is 2nd most common manifestation (after pulmonary disease)

Bimodal distribution
–
Neonatal hepatitis/cholestasis in infants
–
Chronic liver disease/cirrhosis in adults
Microscopic
•
Eosinophilic globules within periportal/periseptal hepatocytes are characteristic

Globules are strongly PAS(+), diastase resistant

Present in periportal/periseptal hepatocytes

Associated histologic features (inflammation, fibrosis) in adults are variable and nonspecific
•
Neonatal hepatitis features cholestasis, hepatocyte injury

Globules may be difficult to detect in young infants
TERMINOLOGY
Abbreviations
Definitions
•
Autosomal recessive genetic disorder characterized by mutations in
SERPINA1 gene

A1AT protein synthesized mainly in liver
–
Major circulating serine protease inhibitor
–
Inhibits neutrophil proteases, thus protecting host tissues from nonspecific injury secondary to inflammation

Mutations result in defective secretion of molecule
–
Most commonly Glu342Lys substitution
–
Protein folds abnormally forming insoluble aggregates instead of being secreted

Deficiency is characterized by emphysema and chronic liver disease
•
Most common inherited metabolic disorder leading to liver transplantation in childhood
•
Most common genetic cause of liver disease in adults and children
ETIOLOGY/PATHOGENESIS
Inherited Metabolic Disorder
•
Highly polymorphic genes with many recognized variants

Variants comprise protease inhibitor (Pi) system
–
Most common variant is PiM

Present in > 90% of USA population

Associated with normal serum A1AT levels
–
Most common deficiency alleles are PiS and PiZ

PiZZ phenotype accounts for most cases of severe A1AT deficiency

∼ 0.5% of population

Typically Caucasians of Northern European ethnicity
–
∼ 2% of individuals are heterozygous for Z allele

Risk of liver disease in heterozygotes is controversial
Accumulation of Mutant Protein
•
Coding sequence defects lead to abnormal polymerization of glycoprotein, preventing export from hepatocyte

Mutant protein accumulates in endoplasmic reticulum of hepatocyte
–
Subsequent decrease in serum A1AT
–
Specific mechanism of hepatocyte injury is unknown
CLINICAL ISSUES
Epidemiology

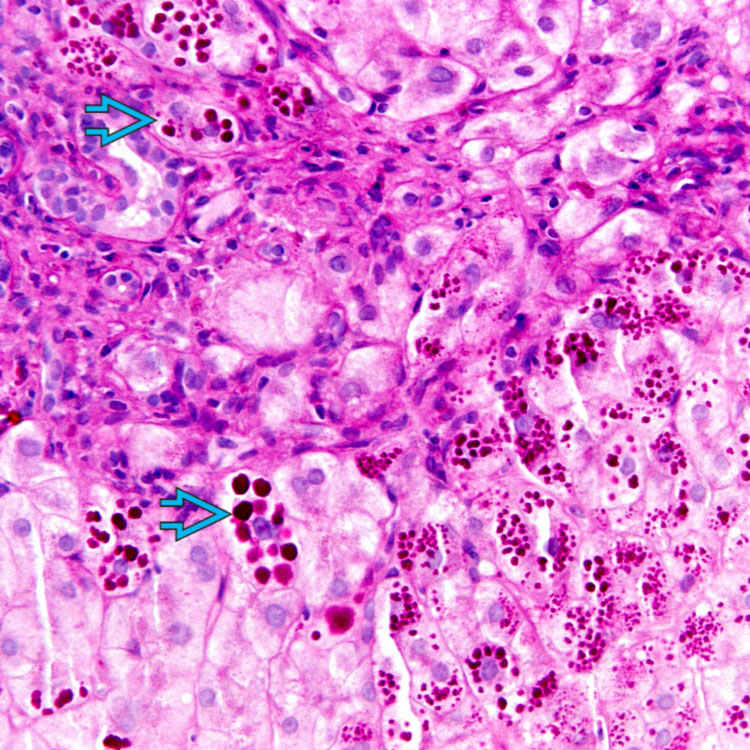
 in periportal areas.
in periportal areas.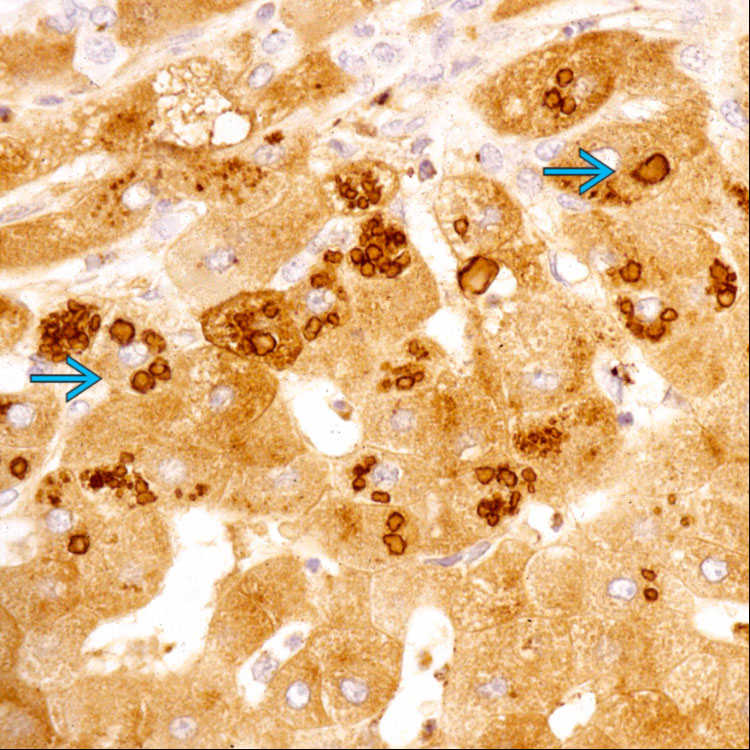
 . The peripheral pattern of staining of each globule is characteristic. In neonates, there is typically more granular cytoplasmic staining, as well-formed globules are not usually present in this age group.
. The peripheral pattern of staining of each globule is characteristic. In neonates, there is typically more granular cytoplasmic staining, as well-formed globules are not usually present in this age group.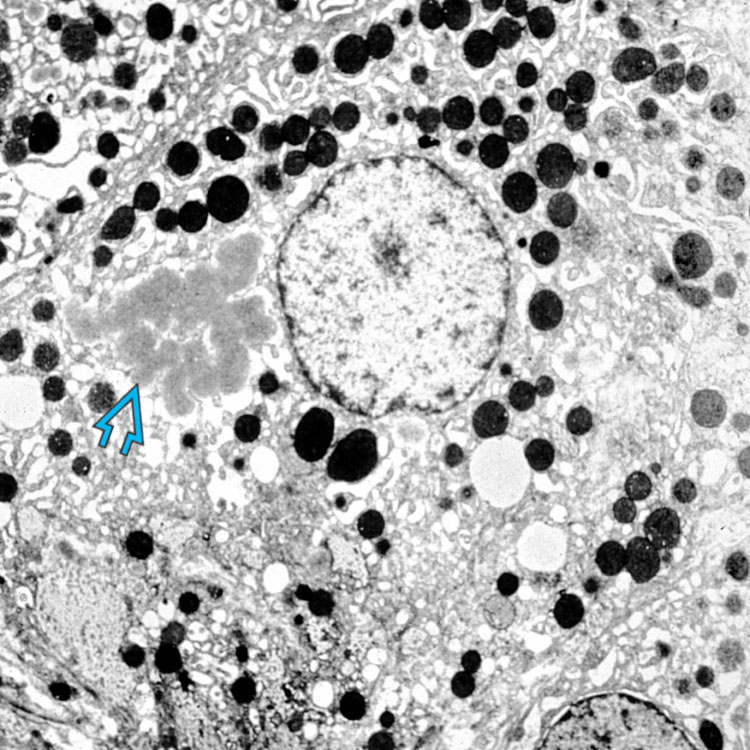
 .
.



