http://evolve.elsevier.com/Edmunds/NP/
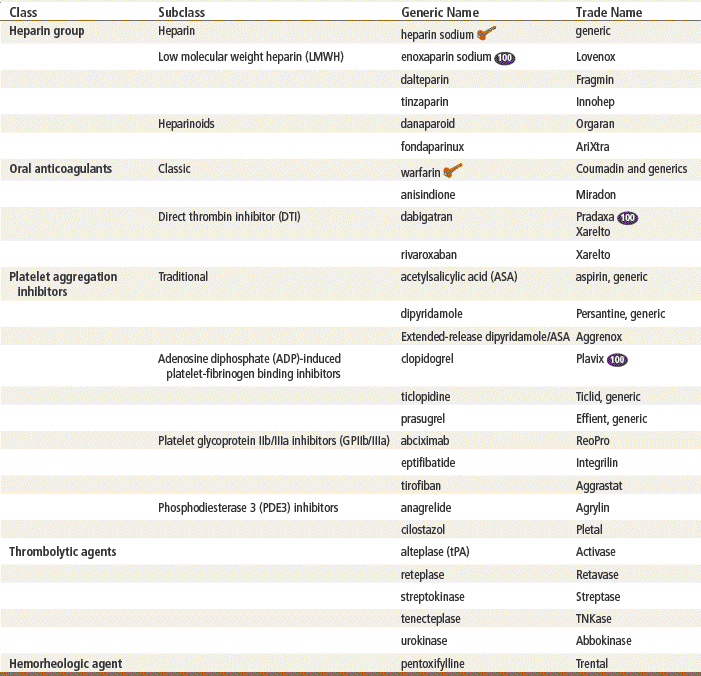
 Top 100 drug;
Top 100 drug;  Key drug.
Key drug.
INDICATIONS
Anticoagulant drugs prolong the body’s ability to form a thrombus (clot) at various points in the coagulation cascade. The goal of therapy is to promote anticoagulation while minimizing hemorrhagic complications through careful monitoring. These medications must be used with great care and often under the supervision of a specialist.
The list of drugs discussed in this chapter is detailed to give the names the provider may recognize. However, only those drugs used in primary care will be discussed in detail. LMWH is more commonly seen now in primary care, although unfractionated heparin (UFH) is still in use. Warfarin, dabigatran, clopidogrel, and aspirin (ASA) are probably the most important drugs in this chapter for the primary care provider. The provider must have a complete understanding of the mechanism of action, dosing, monitoring, and side effects of these drugs in order to prescribe them. The first two groups of platelet inhibitors are seen in primary care. The newer platelet inhibitors remain specialty medications. Thrombolytics and direct thrombin inhibitors (DTIs) are used only in the acute care setting and are included here for those times when the primary provider has contact with patients who are experiencing acute cardiovascular events. Providers need to be aware of the time constraints surrounding initiation of thrombolytic therapy if they are to make timely transfers and referrals to tertiary care centers. The hemorheologic agent is also discussed briefly here. The newest direct thrombin inhibitor, dabigatran (Pradaxa), is discussed in detail.
Therapeutic Overview
A delicate balance must be maintained between the fluidity of the bloodstream and the ability of blood to clot quickly to prevent hemorrhage. In the coagulation system, two pathways are necessary for clotting. The intrinsic clotting pathway is initiated when the blood is exposed to a negatively charged surface such as the in vitro coagulation activators celite, kaolin, or silica. The intrinsic pathway is triggered when blood comes into contact with damaged endothelium or collagen. The extrinsic clotting pathway is triggered by exposure of tissue factor at the site of tissue injury or the addition of thromboplastin to blood. The intrinsic and extrinsic pathways merge on the activation of factor X in what is referred to as the final common pathway (Figure 25-1). Factor X then converts factor II (prothrombin) to IIa (thrombin), and factor IIa converts fibrinogen to a fibrin clot (thrombus).
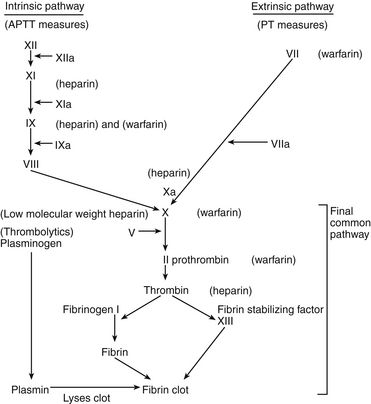
FIGURE 25-1 Coagulation and fibrinogen systems.
Site of action of antithrombotic and thrombolytic drugs.
Platelets provide the initial response to tissue injury (bleeding) and are activated by thrombin, ADP, serotonin, and epinephrine. They next adhere to collagen or to the damaged endothelium and then aggregate to form a platelet plug on the damaged cell wall. During this time, platelets also trigger formation of the active clotting factors VII and X, which leads to clot formation. The fibrin-bound clot framework itself stimulates activation of additional platelets. These platelets release thromboxane A2, serotonin, and ADP, which enhance platelet aggregation and reinforce the formed clot.
Studies suggest that flavonoids found in fruits, vegetables, and some beverages such as tea, coffee, beer, and fruit drinks inhibit several measures of platelet activity such as epinephrine- and ADP-induced GPIIb/IIIa and P-selection expression.
Pathophysiology
The normally protective mechanism can become destructive to the body and can serve as the source of further pathology when clots form in certain areas and prevent tissues from receiving blood. Tissue ischemia and necrosis occur distal to the arterial thrombosis and manifest as MI, stroke, and acute peripheral arterial occlusion. Venous thrombi can result in PE.
A number of acquired factors are associated with increased risk for a thromboembolic event (Box 25-1). Most of these factors are linked to decreased circulation, reduced mobility, or obstruction of blood flow. Many are the result of other diseases or disability or disabilities. One of the critical decisions that a clinician must make is whether it is possible to reduce or control risk factors in order to minimize the chance of thromboembolic events. Clinicians should suspect inherited risk factors, such as deficiency of anticoagulation proteins C and S, antithrombin III deficiency, factor V Leiden mutation, prothrombin G20210A mutation, and factor VIII elevations in patients displaying unusual procoagulable or prothrombotic tendencies. Hyperhomocysteinemia is found in about 5% of the population and is associated with a threefold increase in VTE. Virchow’s triad describes inherited and acquired conditions that place patients at increased risk for developing emboli; these include hypercoagulable states, endothelial injury, and circulatory stasis.
Disease Process
Most of these drugs are used to prevent or treat blood clots that cause thromboembolic events such as stroke, MI, DVT, and PE. The most important pathogenic mechanism in angina and MI is an intracoronary, platelet-rich thrombus on a disrupted, ulcerated, or eroded atherosclerotic plaque leading to partial or complete coronary artery occlusion. The same process occurs in the internal carotid artery or the atria of the heart, and this leads to a stroke. Venous stasis frequently gives rise to clot formation or DVT. If this thrombus dislodges or embolizes, it then causes a PE. PE is the leading cause of preventable hospital death in the United States.
Most cases of chronic peripheral arterial occlusive disease are caused by atherosclerosis. The femoropopliteal, tibioperoneal, aortoiliac, carotid, vertebral, splanchnic, renal, and brachiocephalic arteries are most commonly involved. In chronic arterial occlusive disease, the goals of antithrombotic drug therapy are to relieve symptoms of pain and claudication and to prevent progression of disease that may lead to loss of the limb.
Assessment
Mechanism of Action
Heparin has no effect on existing clots; it prevents or retards formation of new thrombi. Heparin acts at multiple sites in the coagulation system and binds with antithrombin III (AT-III) at two specific sites, resulting in its anticoagulant effect. At the first heparin–AT-III binding site, factor Xa is neutralized, thereby exerting a direct effect on factor X. Factor X is responsible for initiating the final common pathway in the clotting cascade (see Figure 25-1), which ends in clot formation. The second heparin–AT-III binding occurs at the site of conversion of prothrombin to thrombin (factor IIa). With decreased thrombin available, a reduced amount of fibrin is made from fibrinogen. Standard UFH is a mixture of polysaccharide molecules that vary in average molecular weight and composition. Typically, only one third of the molecules in a standard UFH preparation contain the pentasaccharide sequence needed for antithrombin binding and anticoagulation.
Low molecular weight heparin (LMWH) has several advantages over UFH. It has a more predictable anticoagulant effect along with a higher ratio of anti–factor Xa to anti–factor IIa, thus inhibiting the generation of thrombi higher in the clotting cascade. A characteristic of LMWH is that it cannot be monitored with an aPTT as heparin can. Use of LMWH results in a lower incidence of heparin-induced thrombocytopenia and possibly in lower risks of bleeding and osteopenia.
Oral Anticoagulants
Warfarin competitively blocks vitamin K–binding sites and inhibits the synthesis of vitamin K–dependent coagulation factors VII, IX, X, and II (prothrombin) and anticoagulant proteins C and S. Warfarin and anisindione (rarely used) are often referred to as vitamin K antagonists (VKA). At therapeutic levels, warfarin decreases liver synthesis of vitamin K–dependent clotting factors by 30% to 50%. These clotting factors have different half-lives. Factor VII has the shortest half-life (6-9 hr) vs. factors II and X (up to 72 hr). Oral anticoagulants do not reverse ischemic damage or lyse an established thrombus but rather prevent extension of the existing thrombus and the formation of new thrombi by blocking synthesis of clotting factors. Existing clotting factors are not affected; therefore, the onset of action of warfarin is dependent on when existing factors are inactive. This can take several days and should be monitored closely.
It is critical to understand these vitamin K–dependent clotting factors and their half-lives if warfarin is being prescribed. Warfarin is the number one drug causing adverse drug effects in hospitals, sometimes because clinicians do not understand these times. The activity of various clotting proteins (logarithmic scale) is shown in Table 25-2 as a function of time after ingestion of warfarin (10 mg/day po for 4 consecutive days) by a normal subject. Factor VII activity, to which prothrombin time is most sensitive, is the first to decrease. Full anticoagulation, however, does not occur until factors IX and X and prothrombin are sufficiently reduced. Protein C activity falls quickly, and, in some patients, a transient hypercoagulable state may ensue (e.g., coumarin necrosis) (Furie, 2000).
TABLE 25-2
Approximate Half-Life of Vitamin K–Dependent Factors and Anticoagulants
| Factor or Anticoagulant | Half-Life |
| Factor VII | 6 hours |
| Factor IX | 24 hours |
| Factor X | 36 hours |
| Factor II | 50 hours |
| Protein C | 8 hours |
| Protein S | 30 hours |
Compiled from Hoffman R et al, eds: Hematology: basic principles and practice, ed 3, New York, 2000, Churchill Livingstone.
Pradaxa is a new oral anticoagulant, a first-in-class direct thrombin inhibitor. It blocks the activity of thrombin and helps to prevent clot formation. An estimated 2.3 million Americans have atrial fibrillation (AF), and the prevalence is expected to increase 2.5-fold to 5.6 million by 2050, reflecting the growing population of elderly individuals. Approval of Pradaxa was based on the RE-LY (Randomized Evaluation of Long-term Anticoagulant Therapy) trial. This multicenter, multinational, randomized parallel group trial compared Pradaxa (110 mg twice daily and 150 mg twice daily) with open-label warfarin (dosed to target INR of 2 to 3) in patients with nonvalvular, persistent, paroxysmal, or permanent AF with ≥1 risk factors. The primary end point was noninferiority to warfarin in reducing the occurrence of the composite end point, stroke (ischemic and hemorrhagic), and systemic embolism.
A total of 18,113 patients were randomized and followed for a median of 2 years. Pradaxa 150 mg twice daily significantly reduced the primary composite end point of stroke and systemic embolism, as compared with the 110 mg twice daily dose and warfarin. Pradaxa capsules are oral capsules given twice daily and are available in two dosage strengths: 75 mg (for reduced renal function) and 150 mg (for nonrenal compromised patients). There are no requirements for monitoring the INR or other measures for patients taking Pradaxa. When changing from warfarin, it is recommended that Pradaxa be initiated when the INR <2. Pradaxa is the first new oral anticoagulant to be FDA approved in over 50 years. It will compete heavily with warfarin because no INR monitoring is required. However, serious safety concerns exist with Pradaxa because it has a higher risk of GI bleeding in addition to an increased cost.
Platelet Aggregation Inhibitors
Aspirin (ASA) prevents platelet aggregation by inhibiting cyclooxygenase in platelets and endothelial cells, thereby preventing the synthesis of thromboxane A2 and prostacyclin, both of which are potent platelet aggregators and vasoconstrictors.
Dipyridamole increases the body’s adenosine levels, producing vasodilation, particularly of the coronary arteries, which improves blood flow. It also inhibits phosphodiesterase, the enzyme responsible for elevating levels of cyclic adenosine monophosphate (cAMP). Low levels of cAMP are associated with reduced platelet adhesiveness.
Clopidogrel (Plavix) inhibits platelet aggregation by inhibiting the binding of ADP to its platelet receptor and the subsequent ADP-mediated activation of the GPIIb/IIIa complex. The effect is irreversible; platelets exposed to clopidogrel are affected for the remainder of their life span (about 10 days). Ticlopidine inhibits platelet aggregation by altering the function of the platelet membrane to inhibit ADP-induced platelet-fibrinogen binding. The antiplatelet agent prasugrel was approved by the FDA on July 10, 2009. Like clopidogrel and ticlopidine, prasugrel is a platelet inhibitor of the thienopyridine class. All drugs in this class act as ADP receptor antagonists. What makes these drugs unique is their safety profile and pharmacokinetic properties. Although clopidogrel is a widely prescribed agent, it has limitations such as a modest antiplatelet effect, delayed onset of action, and considerable interpatient variability in drug response. All these disadvantages motivated the development of more effective and predictable agents, such as the novel prasugrel. Much controversy has surrounded the approval of prasugrel.
It is uncertain what role this drug will play in the prevention of MI, as well as the optimal dosing and adverse effects profile. Prasugrel is a pro-drug; oxidation by intestinal and hepatic cytochrome P-450 enzymes converts prasugrel into its active metabolite. Prasugrel is rapidly and almost completely absorbed after oral ingestion of a loading dose. Its active form binds irreversibly to the adenosine diphosphate (ADP) P2Y12 receptor on platelets for their life span, thereby inhibiting their activation and decreasing subsequent platelet aggregation. Prasugrel has a greater antiplatelet effect than clopidogrel because it is metabolized more efficiently. Some of the differences in metabolism between clopidogrel and prasugrel may be explained by genetic polymorphisms affecting the cytochrome P-450 system. Prasugrel coadministrated with aspirin is approved by the European Commission for the prevention of atherothrombotic events in patients with acute coronary syndromes (e.g., unstable angina, non-ST segment elevation myocardial infarction, or ST segment elevation myocardial infarction) undergoing primary or delayed percutaneous coronary intervention (PCI). Prasugrel includes a “black box” warning alerting prescribers that the drug can cause significant, and sometimes fatal, bleeding. The drug should not be used in patients with active pathologic bleeding, a history of TIAs (transient ischemic attacks) or stroke, or urgent need for surgery, including coronary artery bypass graft surgery.
Parenteral GPIIb/IIIa inhibitors are used to decrease the rate of ischemic events during balloon angioplasty and to improve coronary patency before stenting. The GPIIb/IIIa receptors are exposed immediately prior to aggregation that allows platelet-to-platelet attachment. This process also requires fibrinogen. The GPIIb/IIIa inhibitor drugs therefore block the binding sites, thereby inhibiting platelet aggregation. These drugs are approved for intravenous use in patients with acute coronary syndrome (ACS) and to prevent restenosis post–percutaneous transluminal coronary angioplasty (PTCA).
Anagrelide (Agrylin) has a mechanism of action that exerts a thrombocytopenic effect and also inhibits platelet aggregation. The thrombocytopenic effect appears to be a result of inhibiting megakaryocyte development in the late, postmitotic stage. In vitro, anagrelide altered maturation stage, size, and ploidy of developing megakaryocytes. Anagrelide does not appear to alter megakaryocyte progenitor cells or mitotic expansion of developing megakaryocyte precursors. The thrombocytopenic effects of anagrelide appear to be specific for humans. Clinically, anagrelide lowers platelet counts and reduces thrombosis, as well as thrombo-hemorrhagic symptoms associated with thrombocythemia.
At dosages higher than those required to produce thrombocytopenia in humans, anagrelide affects platelet aggregation. The drug inhibits platelet aggregation by inhibiting cyclic nucleotide phosphodiesterase and the release of arachidonic acid from phospholipase, possibly by inhibiting phospholipase A2. These actions increase platelet concentrations of cyclic adenosine monophosphate (cAMP). Anagrelide also inhibits collagen-induced platelet aggregation. Resistance to the drug effect does not appear to occur. Anagrelide does not produce significant changes in red cell or white cell counts or in coagulation parameters.
Cilostazol (Pletal) is a quinolone derivative that inhibits PDE3. The pharmacologic effects of cilostazol are multifactorial and include antithrombotic, antiplatelet, and vasodilatory actions. The actions of cilostazol with respect to peripheral arterial disease (PAD) may be particularly significant within the microcirculation. Cilostazol inhibits platelet aggregation caused by ADP, arachidonic acid, collagen, epinephrine, thrombin, and shear stress; it is 10 to 30 times more potent than aspirin in this regard, but cilostazol does not inhibit prostaglandin I2 synthesis. Cilostazol and its metabolites reversibly inhibit platelet aggregation via inhibition of phosphodiesterase (PDE) type 3 activity. This action suppresses degradation of cyclic AMP and increases levels of cyclic AMP in platelets. Cyclic AMP levels are also increased in the vascular tissue, promoting vasodilation. The vasodilatory actions of cilostazol are greater on femoral arteries than on vertebral, carotid, or superior mesenteric arteries. Renal arteries do not vasodilate in response to cilostazol administration.
Thrombolytic Agents
Thrombolytic drugs, also known as fibrinolytics, dissolve blood clots at sites of intravascular injury. They activate tissue plasminogen, which hastens the conversion of plasminogen to plasmin. Enhanced levels of plasmin (a proteolytic enzyme) digest fibrin-bound clots and coagulation factors such as fibrinogen.
Hemorheologic Agents
Pentoxifylline (Trental) decreases blood viscosity, improves erythrocyte flexibility, increases leukocyte deformability, and inhibits neutrophil adhesion and activation. Although the precise mechanism of action is unknown, these actions improve blood flow through microcirculation and increase tissue oxygenation to the affected area. Because of increased reliance on clopidogrel and ticlopidine clinically, the use of pentoxifylline is currently limited.
Treatment Principles
Evidence-Based Recommendations
Treatments for proximal DVT include the following:
Thrombotic treatment in ischemic heart disease includes the following:
Cardinal Points of Treatment
Nonpharmacologic and Pharmacologic Treatment
The ACCP recommends against the use of ASA alone for prophylaxis of VTE. Patients undergoing surgery who are considered to be at moderate to high risk for VTE should receive UFH or LMWH. The use of graduated compression stockings and mechanical methods of thromboprophylaxis, such as intermittent pneumatic compression devices, should be added to the regimen for patients with multiple risk factors for VTE. In addition, all trauma patients with a minimum of one risk factor for VTE, patients admitted to an intensive care unit, those admitted to hospital with CHF or severe respiratory disease, and those who are immobile should receive routine thromboprophylaxis with LMWH and mechanical methods. Patients with confirmed, nonmassive PE are treated with intravenous UFH or subcutaneous LMWH. Patients with biosynthetic valves should receive anticoagulation for 3 months (INR goal, 2 to 3). Long-term prophylaxis for these patients should include ASA (75-100 mg daily), unless AF is present. Patients with ball or caged disk prosthetic heart valves require lifelong anticoagulation (INR goal, 2.5 to 3.5) provided in combination with ASA 75 to 100 mg daily.
Treatment of Confirmed Acute DVT
Short-term treatment with subcutaneous (SC) LMWH, UFH, or fondaparinux for at least 5 days with concurrent initiation of a VKA is recommended. For the initial treatment of an acute DVT, UFH intravenously (IV) with an initial bolus of 80 mg/kg or 5000 units is used. UFH is discontinued when the INR reaches a value >2 for 24 hours. For the first occurrence of DVT with at least one known risk factor, anticoagulation is recommended for 6 months. DVT that is idiopathic is treated for 6 to 12 months with anticoagulation, with a target INR of 2 to 3.
Treatment of PE
Patients with stable PE are treated with UFH and warfarin anticoagulation. LMWH is beneficial in submassive PE and DVT but remains controversial for the treatment of massive PE. In those with hemodynamically unstable and life-threatening PE, thrombolytic therapy is the treatment of choice. Intravenous alteplase is the preferred agent.
Prevention of Stroke in Patients with Ischemic or Transient Ischemic Attack
The ACCP and The American Stroke Association Council on Stroke recognize that patients with a history of cardiac and/or cerebral vascular disease have an increased risk of recurrent stroke. Those with high-risk sources of cardiogenic embolism such as AF, chronic or paroxysmal, should be anticoagulated. In addition, systemic embolism or stroke occurs in approximately 12% of acute MI patients, particularly when complicated by left ventricular thrombus. ASA and VKA anticoagulation is recommended for this group.
Treatment of Acute Ischemic Stroke
For selected patients experiencing acute ischemic stroke (i.e., meeting the NINDS trial criteria), IV recombinant tissue plasminogen activator (e.g., rtPA, alteplase) should be initiated as quickly as possible; within 3 hours of symptom onset is recommended. The recommended dose of IV rtPA is 0.9 mg/kg, with maximum dose of 90 mg. This recommendation has not changed from previous recommendations. Alteplase is contraindicated if onset of symptoms occurred 3 hours previously. Patients not receiving thrombolytics should receive early ASA therapy (150-325 mg daily). The use of streptokinase or full-dose anticoagulation with IV or SQ UFH or heparinoids in acute ischemic stroke is discouraged, as it is associated with an increased risk of bleeding complications. Currently, the administration of anticoagulation or antiplatelet agents during the first 24 hours after treatment with IV rtPA for acute ischemic stroke is contraindicated. The oral administration of aspirin (325 mg) within 24 to 48 hours after stroke onset is recommended, and a small but significantly significant decrease in mortality and mortality has been demonstrated. Although the combination of clopidogrel and aspirin is indicated in acute coronary syndromes, this combination has not been studied in acute ischemic stroke and is not recommended. In patients who have had a cardioembolic stroke caused by nonvalvular AF, heparin anticoagulation as primary therapy does not reduce the end points of death and disability.
In patients with extracranial or vertebral atherosclerosis who had an ischemic stroke or TIA, treatment with aspirin alone (75-325 mg), clopidogrel (75 mg daily), or the combination of aspirin or extended release dipyridamole (25 mg and 200 mg twice daily, respectively) is recommended.
Treatment of AF and Prevention of Stroke in AF
Persistent and paroxysmal AF is a predictor of first stroke and recurrent stroke risk. Those with persistent or paroxysmal AF and prior ischemic stroke, TIA, systemic embolism, or aged ≥75 years benefit from warfarin anticoagulation at an INR goal of 2.5 (range 2.0 to 3.0). For AF patients with more than one moderate risk factor (e.g., aged 75 years, hypertension, impaired LV function, ejection fraction 35%, or diabetes mellitus), warfarin anticoagulation is recommended. For patients unable to take VKAs, aspirin alone is recommended. The use of clopidogrel plus aspirin is not recommended due to risk of hemorrhage. The evidence supporting the efficacy of ASA is weaker than it is for warfarin. The Stroke Prevention in Atrial Fibrillation Trial used ASA at a dose of 325 mg daily. However, the results of the trial indicate the best efficacy and safety profile at a dose of 75 to 100 mg daily. When mitral stenosis is present or a prosthetic valve is in place, warfarin is indicated. The INR should be maintained at 2.5 for those with rheumatic valve disease (range 2.0 to 3.0) and INR of 3.0 (range 2.5 to 3.5) for mechanical prosthetic valves. ASA may be added to the regimen. Warfarin therapy of 3 to 4 weeks’ duration is recommended before electrical cardioversion of new onset AF (48 hours) is attempted. If onset of AF has occurred within 48 hours, cardioversion can be done without anticoagulation. In patients with rheumatic mitral valve disease and AF or a history of systemic embolism, anticoagulation plus ASA (75-100 mg daily) is indicated. If unable to tolerate ASA, clopidogrel is used. For patients with mitral valve prolapse (MVP), but without a history of systemic embolism or TIA, antithrombotic therapy is not recommended. If these conditions coexist with MVP, ASA (75-100 mg daily) is suggested.
Antithrombotic Treatment for ACS
Patients with non–ST-segment elevation (NSTE) ACS should receive ASA 162 to 325 mg po (chewed) immediately. When patients with ACS continue to exhibit symptoms after 1 hour of conventional therapy, abciximab or eptifibatide should be started. In patients who will not undergo diagnostic cardiac catheterization within 5 days of the event, bolus clopidogrel (300 mg) is recommended, followed by 75 mg daily for 9 to 12 months, in addition to ASA. If angiography is performed immediately, clopidogrel 300 mg should be given 6 hours prior to the procedure if possible. In patients considered at moderate to high risk for future events, eptifibatide or tirofiban therapy is encouraged, in addition to ASA and heparin.
Aspirin (81-162 mg daily) is recommended for all patients with coronary artery disease. Clopidogrel (75 mg daily) is used as an alternative for those few patients unable to take aspirin. Warfarin should not be given with clopidogrel and ASA concurrently.
Thrombolytic Therapy for ACS
For patients who are experiencing ischemic symptoms of <12 hours’ duration with ST-segment elevation or new left-bundle branch block, a fibrinolytic drug should be given (alteplase, reteplase, or tenecteplase may be used). If symptom duration is <6 hours, the preferred drug is alteplase. In the acute treatment of NSTE ACS, LMWH rather than UFH is considered standard therapy. Contraindications to thrombolytic use include any history of intracranial hemorrhage, closed head trauma, or ischemic stroke within the previous 3 months. All patients without ASA allergy should be given ASA 162 to 325 mg at the time of initial evaluation, regardless of fibrinolytic therapy. Clopidogrel is given if ASA allergy is known.
Therapy in Peripheral Arterial Occlusive Disease
Patients with disabling intermittent claudication who are not candidates for surgery or catheter-based intervention should be treated with cilostazol rather than pentoxifylline. Anticoagulation is not recommended for patients with intermittent claudication. Those with chronic limb ischemia require lifelong ASA therapy. In patients with acute arterial emboli or thrombosis, the use of UFH followed by lifelong warfarin anticoagulation is recommended. Patients with asymptomatic or recurrent carotid stenosis require lifelong ASA therapy.
Use of Antithrombotic Drugs During Pregnancy
For women requiring long-term anticoagulation with warfarin and considering pregnancy, UFH or LMWH should be substituted for warfarin as soon as pregnancy is achieved. Pregnant women with acute VTE are treated with adjusted-dose LMWH throughout the pregnancy and for at least 6 weeks postpartum. Alternatively, IV UFH may be given for 5 days, followed by LMWH for the duration of the pregnancy.
Heparin
UFH is administered as a continuous intravenous infusion on an inpatient basis. For the prevention of VTE, a dose of 5000 units is given every 8 or 12 hours postoperatively. For the treatment of patients with VTE or ACS, a loading dose of ≈80 units/kg is followed by an infusion of 9 to 18 units/kg/hr, depending on the institution. Begin to monitor aPTT ≈3 to 4 hours after initiation so that any necessary adjustments to the dosage can be made. Generally, treatment is discontinued when the patient is past the risk of thromboembolic complications, but this varies with the specific indication. UFH is most often administered intravenously for 5 to 10 days. If indicated, oral anticoagulation with warfarin is given in conjunction (overlapping) with the heparin infusion because the maximal therapeutic effect of warfarin is not fully reached until after 4 to 5 days (Figure 25-2). Heparin resistance is seen in febrile patients and in those with active thrombosis, phlebitis, infection, MI, cancer, or heparin-induced thrombocytopenia (HIT). Concern has arisen about the possibility of increasing the risk of osteoporosis with prolonged standard heparin administration (>3 months, at an accumulated dose of 20,000 units).
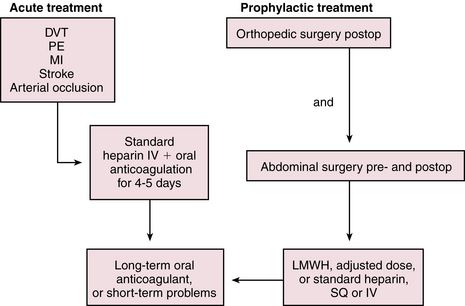
Low-dose UFH or LMWH is given subcutaneously for prophylaxis of DVT, which may lead to PE in susceptible patients. LMWHs (e.g., enoxaparin) and the anti-Xa inhibitor fondaparinux are the preferred treatments for prevention of VTE in patients undergoing major orthopedic surgery such as total hip or knee replacement or hip fracture surgery. UFH, LMWH, or fondaparinux is preferred for major abdominal surgery and for those at risk for DVT or PE because of severely restricted mobility during an acute illness. Use of LMWH as bridge therapy has increased and is indicated for anticoagulated patients removed from warfarin prior to surgery, for AF patients following percutaneous coronary intervention, and for patients discharged on warfarin with a subtherapeutic INR.
A clinically devastating complication of heparin therapy is immune-mediated HIT, which is seen in ≈0.3% to 3% of patients. Although thrombocytopenia is associated with bleeding complications, HIT is uniquely associated with thrombosis. The rate of cross-reactivity between UFH and LMWH is 80% to 100%; therefore, if continued anticoagulation is needed in a situation in which UFH has been used, LMWH cannot be used. DTIs (e.g., argatroban, lepirudin), as well as fondaparinux, may be used in this setting. Warfarin may be prescribed for long-term use in patients with HIT.
Oral Anticoagulants
Oral anticoagulation therapy is recommended for long-term use in patients with the following conditions: persistent or paroxysmal atrial fibrillation, acute MI and LV thrombus, cardiomyopathy, rheumatic mitral valve disease, mechanical prosthetic heart valves and bioprosthetic heart valves with no other source of thromboembolism, DVT, and PE. In addition, prophylactic anticoagulation therapy is recommended for patients considered at high risk for venous or arterial thromboembolism. ASA and anticoagulation are not recommended following acute ischemic stroke. Studies have failed to show an improvement in outcomes with anticoagulation use.
Warfarin therapy often is started concurrently with inpatient heparinization. Warfarin therapy is then continued as short-term adjunct therapy in DVT, post-MI, in PE, and after total joint replacement for up to 3 months. It is used indefinitely for recurrent DVT, PE, chronic AF, and post–embolic stroke, and with cardiomyopathy, left ventricular aneurysm, valvular heart disease, and mechanical cardiac valve replacement. It may be started on an outpatient basis without pretreatment with heparin. No consensus has been reached with regard to perioperative management of patients on warfarin or the INR level that must be reached before surgery can be safely performed.
Loading doses are not necessary and can precipitate thrombotic events in rare cases through rapid depletion of proteins C and S. It is recommended that patients begin with 5 mg once daily (2.5 mg for elderly) for 5 to 7 days and then adjust according to INR. Minor adjustments then can be made because warfarin is available in 1-, 2-, 2.5-, 3-, 4-, 5-, 6-, 7.5-, and 10-mg dosing strengths. In clinical practice, however, the provider will see patients who are taking warfarin in accordance with unusual dosing schedules (e.g., 5 mg q Tues., Thurs., Sat., and 7.5 mg q Wed., Sun., etc.). This practice arises from prescriber misunderstanding of warfarin pharmacokinetics and dynamics. There is no clinical reason for this, and it adds to poor patient compliance. When a patient is initiated on warfarin therapy, he or she should choose between brand or generic and remain on it. However, choosing a generic can complicate monitoring because many generics are available. Patients who are taking branded warfarin, Coumadin, should not use generic warfarin interchangeably because of differing bioavailability factors and the need to maintain a steady state.
Excessive warfarin anticoagulation is commonly overcorrected. Only in cases of an INR >9, life-threatening bleeding, or emergent surgery does warfarin require high-dose (5-10 mg) vitamin K. Excessive doses of vitamin K can create warfarin resistance and can place the patient at increased risk of thrombosis. In most cases, it is recommended that warfarin be stopped temporarily when INR is >5 and hemoglobin is stable (Table 25-3). Determine the source of excessive anticoagulation; possibilities include medication administration error, dietary indiscretion, alcohol overuse, acute febrile illness, diarrhea, new prescription or OTC medications, new herbal remedies, and vitamin supplements.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


